
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
August 2025
1: Systematic Analysis of Cellular Cross-Talk Reveals a Role for SEMA6D-TREM2 Regulating Microglial Function in Alzheimer’s Disease
2: Single-Cell Transcriptomic Landscape of the Neuroimmune Compartment in Amyotrophic Lateral Sclerosis Brain and Spinal Cord
3: Alpha-Synuclein Abundance and Localization are Regulated by the RNA-Binding Protein PUMILIO1
4: The Role of Astrocytes in Human Huntington's Disease Pathology
2: Single-Cell Transcriptomic Landscape of the Neuroimmune Compartment in Amyotrophic Lateral Sclerosis Brain and Spinal Cord
3: Alpha-Synuclein Abundance and Localization are Regulated by the RNA-Binding Protein PUMILIO1
4: The Role of Astrocytes in Human Huntington's Disease Pathology
 |  | |
| Gina Finan, PhD | Tae-Wan Kim, PhD |
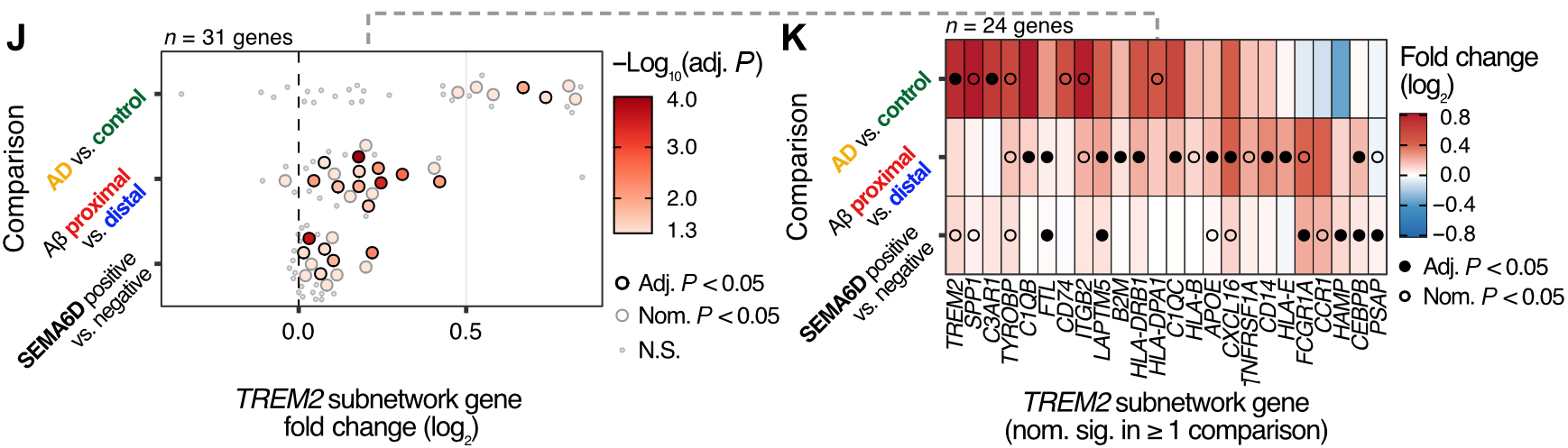
In work recently published in Science Translational Medicine, we set out to investigate how brain cell types communicate with one another and how these interactions become disrupted in Alzheimer’s disease (AD). By integrating single-nucleus RNA sequencing with spatial transcriptomics, we identified a previously unrecognized interaction between neuronal SEMA6D and microglial TREM2.
We found that the SEMA6D–TREM2 signaling pathway is activated around amyloid plaques and enhances the ability of microglia to clear toxic Aβ aggregates. Importantly, as the disease progresses, the loss of SEMA6D expression is associated with diminished microglial activation and impaired plaque clearance, suggesting a mechanism that may contribute to worsening pathology at later stages.
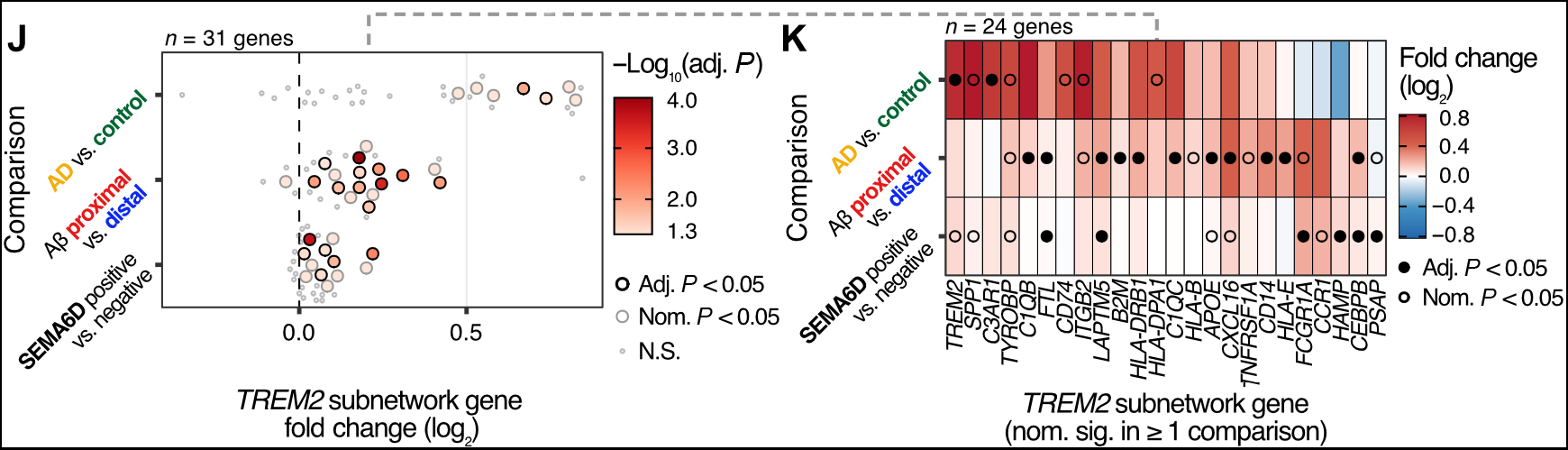 Figure 2 (J&K). Changes in gene expression in the TREM2 cross-talk subnetwork associated with disease status, Aβ plaque proximity, and presence of SEMA6D-expressing cells.
|
It was especially gratifying to see this pathway emerge from computational prediction, be confirmed in human brain tissue, and then validated in functional experiments using stem cell–derived microglia in a 3D triculture AD model. This work highlights how bridging large-scale data analyses with experimental systems can uncover new insights into disease biology and point to potential therapeutic targets.
This work was carried out in close collaboration with Dr. Oscar Harari’s lab at The Ohio State University.
Gina Finan, PhD
Associate Research Scientist in the Department of Pathology and Cell Biology
gmf6@cumc.columbia.edu
Tae-Wan Kim, PhD
Associate Professor of Pathology and Cell Biology (in the Taub Institute)
twk16@cumc.columbia.edu
 |  |  | ||
| John Tuddenham, MD, PhD | Vilas Menon, PhD | Marta Olah, PhD |
In a multi-institutional collaborative study, we recently explored how amyotrophic lateral sclerosis (ALS) affects the neuroimmune compartment. Utilizing the ALS tissue donation program of Target ALS and the Eleanor and Lou Gehrig ALS Center (both directed by Dr. Neil Shneider at CUIMC), we isolated live microglia from ALS brain and spinal cord autopsy specimens and profiled their transcriptomes using single cell RNA-sequencing. We characterized the composition, relative abundance, and disease association of several microglia and non-microglia immune cell phenotypes across multiple brain regions along the neuraxis in ALS.
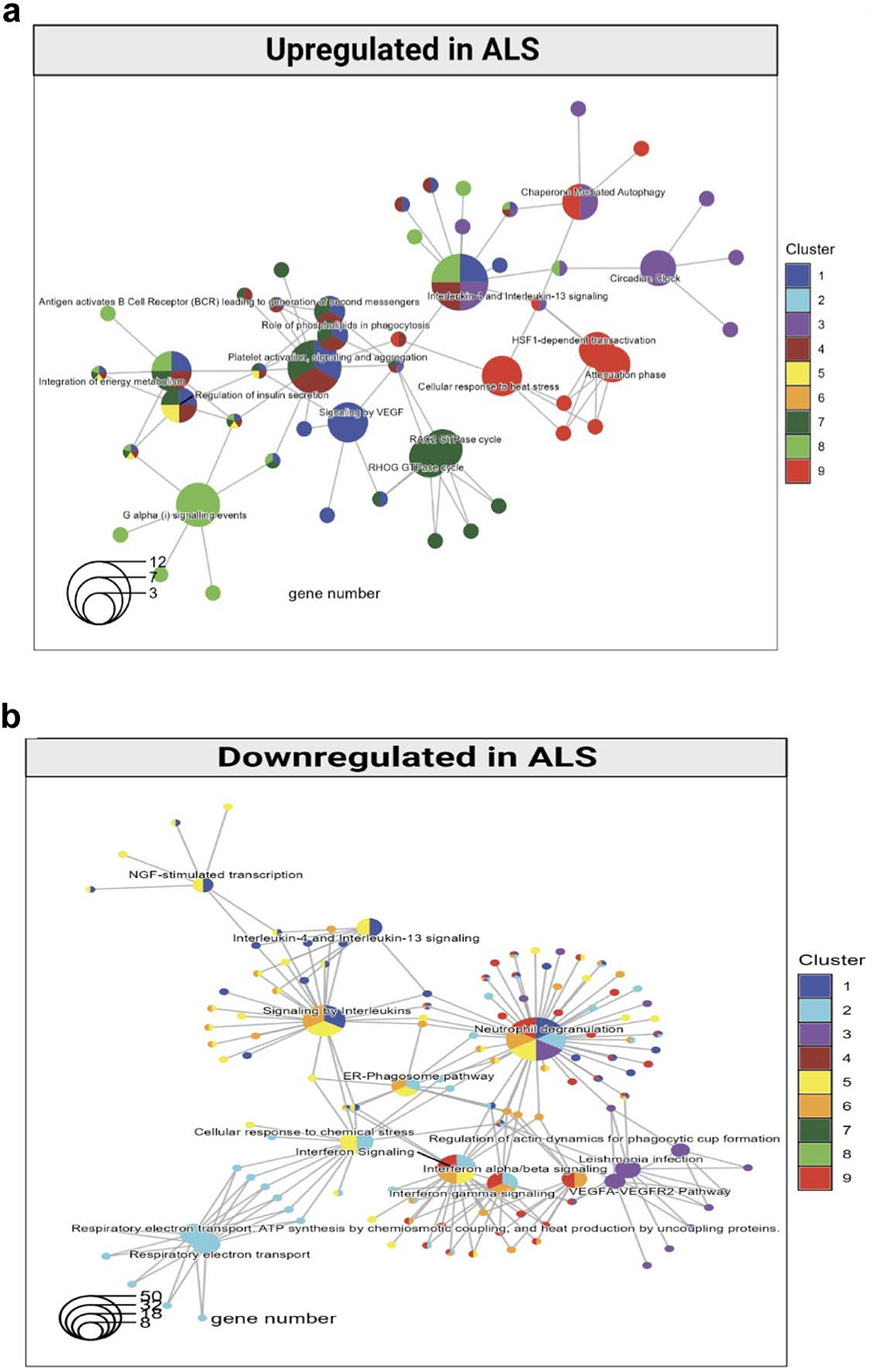
Figure 3 (a&b). ALS results in functional changes in microglia subsets.
As recently reported in Acta Neuropathologica, this approach uncovered the ALS specific expansion of MG2 microglia—an undifferentiated subtype marked by dysregulated respiratory electron transport—and enrichment of interferon-response microglia (MG4) in cases of rapidly progressing ALS. Potentially protective microglial subsets were notably depleted. Functional analyses revealed upregulation of stress, inflammatory, and metabolic pathways in ALS-associated microglia, and implicated SON as a key transcriptional regulator driving these shifts. In addition to microglial changes, the authors observed increased presence of peripheral immune populations, including T cells and NK cells, particularly in the spinal cord.
These findings add to growing evidence supporting the need to understand disease specific dysregulation of the neuroimmune compartment in human neurodegenerative diseases. Our study provides a high-resolution atlas of microglia (and non-microglial immune cell) phenotypes in ALS and offers new targets for microglia-focused therapeutic strategies.
Marta Olah, PhD
Assistant Professor of Neurological Sciences (in Neurology and the Taub Institute)
mo2738@cumc.columbia.edu
Alpha-Synuclein Abundance and Localization are Regulated by the RNA-Binding Protein PUMILIO1
 |  | |
| Ulrich Hengst, PhD | Vincenzo A. Gennarino, PhD |
At the core of many neurodegenerative diseases, including Alzheimer’s and Parkinson’s disease, is a breakdown of proteostasis, the cellular process that ensures that proteins are expressed at the right amount within neurons. The abundance of proteins within cells can be affected by several mechanisms that control the production of individual proteins (transcription, translation) or conversely their removal or degradation. In the context of neurodegenerative diseases, the latter mechanisms have arguably received most attention, but more recently, it has emerged that dysregulation of transcriptional and translational mechanisms can likewise be pathogenic.
Figure. The relative abundance of α-synuclein mRNA and the RNA-binding protein PUM1 controls the expression of α-synuclein. In Parkinson’s disease, α-synuclein is overabundant, for example, due to a reduction of the repressive effects of PUM1. Increasing PUM1 activity normalizes α-synuclein expression in these cases.
In collaboration with the laboratory of Dr. Vincenzo Gennarino in the Department of Genetics and Development, my lab at the Taub Institute contributed to a new multidisciplinary study investigating the post-transcriptional control of α-synuclein expression. In Parkinson’s disease (PD) and other synucleinopathies, α-synuclein accumulates, but the reasons for this remain unclear. Published in Cell Reports, our findings demonstrate that an RNA-binding protein, PUM1, binds to the 3′ untranslated region of α-synuclein mRNA. In induced neurons from patients with an α-synuclein locus triplication, PUM1 mRNA levels are lower compared to healthy controls. However, increasing PUM1 levels normalizes α-synuclein mRNA and protein levels. Furthermore, in microfluidic chamber experiments, silencing PUM1 caused a redistribution of α-synuclein mRNA between the soma and axons. The suppressive effect of PUM1 on α- synuclein was at least partially mediated by the microRNA mir-7.
The relevance of these findings for neurological disorders is further emphasized by the finding that several individuals with PD in clinical databases carry variants in PUM1 that affect its RNA-binding ability. Therefore, the results of this study will help us understand how RNA-binding proteins regulate α-synuclein and could inform the development of new therapies for synucleinopathies.
Ulrich Hengst, PhD
Professor of Pathology & Cell Biology (in the Taub Institute)
uh2112@cumc.columbia.edu
The Role of Astrocytes in Human Huntington's Disease Pathology
|  | |
| Osama Al-Dalahmah, MD, PhD | James E.Goldman, MD, PhD |
Huntington’s disease (HD)is a dominantly inherited disorder of abnormal movements and cognitive loss caused by an increased number of CAG triplet repeats in the HTT gene. Based on published studies of astrocyte gene expression in human Huntington’s disease from our colleagues and us, using single nuclei RNA sequencing from frozen tissues from the New York Brain Bank, we were asked to write a review for the Journal of Huntington’s Disease on astrocytes in human Huntington’s Disease. There are a number of mouse models of HD in which astrocytes have been examined, but many fewer reports of astrocytes in the human brain. When most neuroscientists think of astrocytes, they think of the “protoplasmic” astrocytes, found in gray matter all over the central nervous system, and are involved in ion and neurotransmitter homeostasis, among other functions. But there are other astrocyte types, including the “fibrous-like” astrocytes of white matter and subpial and subependymal areas, which have very different morphologies and in which we have found different gene expression patterns.
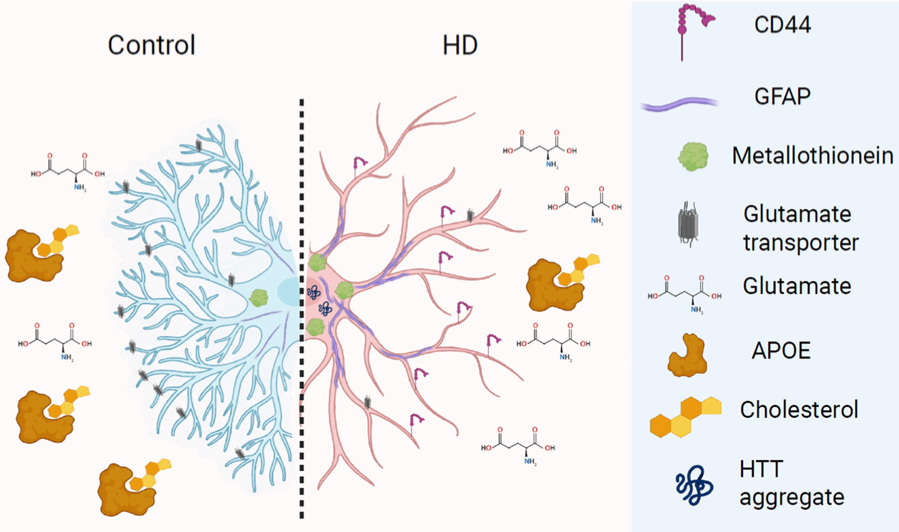
Figure 2. Summary of changes in HD astrocytes.
Taken from Osama Al-Dalahmah and James E. Goldman, “The role of astrocytes in human Huntington’s disease pathology” J Huntington’s Disease 2025; 14:214-228.
In HD, although all cells express the mutant HTT, the medium spiny neurons of the striatum show the most severe degeneration. But other areas, including the cerebral cortex and white matter also show neuropathology, although to a lesser extent. We have examined gene expression in astrocytes in several regions of the HD brain: the cerebral cortex, the subcortical white matter, the cingulate cortex, the caudate nucleus, and the nucleus accumbens. We found several dramatic changes. 1. Many of the “protoplasmic” astrocytes have lost their normal gene expression patterns, and in particular transporters for glutamate and potassium ion channels, and have acquired patterns that look more like those of “fibrous-type” astrocytes, including the acquisition of CD44, a cell surface matrix receptor characteristic of fibrous astrocytes. 2. The protoplasmic astrocytes of the cortex upregulate metallothionein genes, which we believe are neuroprotective, being able to chelate several heavy metals, which are increased in the HD brain. Caudate astrocytes appear less able to do this. 3. HD causes changes in astrocyte lipid metabolism. For example, in our lipidomics analysis, we find increases in long chain fatty acids, which in vitro sensitize neurons to oxidative death. Astrocytes alter levels of transcripts for fatty acid desaturation and elongation. 4. Astrocytes contain HTT aggregates, as shown by others, similar to those in neurons. 5. Astrocytes do not show significant somatic expansion of the CAG repeats, which is in contrast to the expansions found in neurons.
We think of these changes in astrocytes as a continuum of pathological “states” from the quiescent protoplasmic cells to the fibrous-like cells, and with the changes of state, dramatic changes in gene expression. We are now working with Rafe Batchelor and Vilas Menon in Neurology to compare astrocytes, and other glia, in Alzheimer’s, Parkinson’s and HD, to look for both similar and different changes in astrocyte gene expression in these different neurodegenerative disorders.
Osama Al-Dalahmah, MD, PhD
Assistant Professor in Pathology and Cell Biology
oa2298@cumc.columbia.edu
James E.Goldman, MD, PhD
Professor of Pathology and Cell Biology (in Psychiatry)
jeg5@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.