
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
July 2015
» #1 Candidate genes for Alzheimer's disease are associated with individual differences in plasma levels of beta amyloid peptides in adults with Down syndrome.
» #2 Gene-Wise Association of Variants in Four Lysosomal Storage Disorder Genes in Neuropathologically Confirmed Lewy Body Disease.
» #2 Gene-Wise Association of Variants in Four Lysosomal Storage Disorder Genes in Neuropathologically Confirmed Lewy Body Disease.
 |  |  |
| Nicole Schupf, PhD | Lorraine Clark, PhD | Joseph H. Lee, DrPH |
Amyloid β (Aβ) plays a critical role in the development of Alzheimer's disease (AD). Amyloid β peptides Aβ40 and Aβ42 are the two major species generated from the amyloid precursor protein (APP). Brain levels of Aβ42 increase early in the development of dementia, and elevated plasma Aβ42 levels have been proposed as a risk factor related to both age at onset and risk of AD.
Dr. Nicole Schupf and colleagues have been studying the role of plasma Aβ peptides in risk for AD in adults with Down syndrome (DS). Individuals with DS have increased risk for AD neuropathology and clinical dementia, which has been attributed to having three copies of the gene for APP on chromosome 21, which is overexpressed and leads to high levels of beta amyloid. They have found that high initial levels and subsequent declines in plasma Aβ42 and the Aβ42/Aβ40 ratio are associated with earlier onset and increased risk for AD. However, there are large individual differences in initial Aβ peptide levels and a wide range of age at onset of AD within this population, suggesting a more complex underlying mechanism and a role for additional risk factors.
The factors that influence individual differences in plasma Aβ peptides are not well understood. Genetic risk factors may influence the development of AD by increasing production of Aβ or by reduced clearance or excess deposition of Aβ. Multiple genome-wide studies have identified potential genetic pathways for AD but only a few studies have examined their relation to Aβ peptide levels. Dr. Schupf and colleagues reasoned that individuals with DS may be a population group with increased sensitivity for revealing such pathways. Recently published in Neurobiology of Aging, Drs. Schupf, Clark and Lee, with collaborators from Taub, the NYS Institute for Basic Research in Developmental Disabilities, and the Kennedy Krieger Institute examined the relation of candidate genes for AD to baseline levels of Aβ42, Aβ40 and the Aβ42/Aβ40 ratio in older adults with DS.
Participants were 254 non-demented adults with Down syndrome, 30-78 years of age. Candidate genes included the top candidate genes from the ALZGENE database and additional positional candidate genes from published genome wide linkage and association studies. Genomic DNA was genotyped using an Illumina GoldenGate custom array to evaluate trisomies. The investigators used multivariate linear regression to examine differences in levels of Aβ peptides associated with the number of risk alleles, adjusting for co-factors. For Aβ42 levels, the strongest gene-wise association was found for a SNP on CAHLM1; for Aβ40 levels the strongest gene-wise associations were found for single nucleotide polymorphisms (SNPs) in IDE and SOD1, while the strongest gene-wise associations with levels of the Aβ42/Aβ40 ratio were found for SNPs in SORCS1. Broadly classified, variants in these genes may influence APP processing (CALHM1, IDE), vesicular trafficking (SORCS1), and response to oxidative stress (SOD1). These findings support the hypothesis that individual differences in Aβ processing or deposition, distinct from overexpression of APP, may act as an initial step in the pathogenesis of AD. Schupf et al. are currently examining the relation of SNPs in candidate genes to change in Aβ peptides, and to age at onset and risk for AD. The findings from their studies will allow them to better understand genetic factors that contribute to amyloid and other pathways involved in neurodegeneration.
Nicole Schupf, PhD
Professor of Epidemiology (in the Gertrude H. Sergievsky Center, in the Taub Institute, and in Psychiatry)
Ns24@cumc.columbia.edu
Lorraine Clark, PhD
Associate Professor of Pathology and Cell Biology (in the Taub Institute)
Lc654@cumc.columbia.edu
Joseph H. Lee, DrPH
Associate Professor of Epidemiology (in the Gerturde H. Sergievsky Center and Taub Institute)
Jhl2@cumc.columbia.edu
| |
| Lorraine Clark, PhD | Joseph H. Lee, DrPH |
Dr. Lorraine Clark has had a longstanding interest in the role of the lysosomal and autophagy pathway in Parkinson's disease and dementia with Lewy Bodies. Together with her colleagues in the departments of Neurology, Pathology and Cell Biology, the Taub Institute, and the Gertrude H. Sergievsky Center, the Clark Laboratory has contributed to the identification of genetic factors in these pathways that are risk factors for Parkinson's disease and dementia with Lewy Bodies. Interestingly, many of the Parkinson's disease genes identified to date, such as LRRK2, GBA (Gaucher disease), SMPD1 (Niemann Pick, SNCA, PARK2, PARK16, PINK1, ATP13A2 (lysosomal ATPase), VPS35, SCARB2 (lysosomal protein) are involved in the autophagy-lysosomal pathway and collectively these studies point to a pathway that could be targeted for therapeutic intervention.
In 2005, Dr Clark's lab was awarded funding from the NIH (R21 NS050487) to investigate the role of the Glucocerebrosidase (GBA) gene as a risk factor for Parkinson's disease in the Ashkenazi Jewish population. Gaucher disease (GD) is the most common inherited lysosomal storage disorder in humans and is caused by mutations in the gene encoding the lysosomal enzyme Glucocerebrosidase. These studies demonstrated that the GBA gene is a susceptibility gene for Parkinson's disease in both Jewish and non-Jewish populations and that individuals heterozygous for a GBA mutation are at risk of developing Parkinson's disease and at an earlier age at onset. Subsequent studies that Dr Clark's lab have contributed to have demonstrated that GBA is one of the most significant risk factors for Parkinson's disease identified to date and is also a risk factor for neuropathologically confirmed dementia with Lewy bodies.
In 2009, Dr Clark's lab was awarded funding from the NIH (R01 NS060113) to continue her studies of GBA in addition to studying the role and mechanism of other lysosomal storage disorder genes in Parkinson's disease and dementia with Lewy Bodies. These studies have focused on the genes that cause four of the most common lysosomal storage disorders in the Ashkenazi Jewish population that include Gaucher disease (GD), Tay Sachs Disease (TSD), Niemann Pick Disease, and Mucolipidosis type IV (MLIV).
The Lysosomal storage diseases (LSDs) are a group of metabolic disorders caused by genetic mutations in three classes of proteins: 1) lysosomal hydrolases required for catabolic degradation, 2) lysosomal membrane proteins important for catabolite export or membrane trafficking and 3) non-lysosomal proteins that indirectly affect lysosomal function. The importance of the lysosome is highlighted by the large number of diseases that have been documented ranging from cancer to neurodegenerative disease, and the pathology in several tissues and organs. The classic feature of LSDs is the accumulation of undigested lipids in the lysosome leading to lysosomal dysfunction and cell death. More than 60 LSDs have been described which are multisystemic and clinically heterogeneous but often have a neurological involvement. A spectrum of neurological manifestations has been noted in patients with four of the most common lysosomal storage disorders in the AJ that include Gaucher disease (GD), Tay Sachs Disease (TSD), Niemann Pick Disease and Mucolipidosis type IV (MLIV) that Dr Clark's lab has studied and that can be classified based on age at onset and severity of the disease. The extent of variation of nervous system involvement and correlation with specific lysosomal gene mutations in patients with an intermediate or mild late-onset phenotype is currently an active area of research. However, a spectrum of neurological manifestations has been noted in patients with mutations in each of these genes ranging from epilepsy, myoclonus, supranuclear gaze palsy, cerebellar ataxia, psychiatric symptoms, parkinsonism and dementia. Further evidence that the genes that cause LSDs may also be involved in Parkinson's disease and dementia with Lewy bodies comes from studies of the neuropathology of several lysosomal storage disorders. a-Synuclein immunoreactivity, including Lewy bodies, which is the hallmark of the neuropathology of Parkinson's disease and Dementia with Lewy Bodies has been described as a predominant feature seen in the neuropathology of several lysosomal storage disorders, including notably Gaucher disease, Sandhoff disease, Tay Sachs disease, and Sanfilippo syndrome among other. Subjects with GBA mutations who develop PD demonstrate on autopsy typical neuropathological hallmarks with post mortem a-synuclein post mortem inclusions and a-synuclein aggregates in neuronal cells.
In a recent paper published online in PLOS ONE, Drs. Clark, Lee, and colleagues have shown that variants in other lysosomal storage disorder (LSD) genes in the same pathway as GBA are associated with Lewy bodies. In this new genetic study, four LSD genes, GBA, HEXA, SMPD1 and MCOLN1, were examined in 231 brain autopsies from the New York Brain Bank at Columbia University. The functional effect of GBA mutations was also determined by performing a biochemical analysis, including enzymatic activity and lipidomic analysis, in a subset of brains. Their study indicates that variants in GBA, SMPD1 and MCOLN1 are associated with Lewy body pathology. Biochemical data comparing GBA mutation carriers to non-carriers shows that haploinsufficiency or partial enzyme activity can lead to significant changes in sphingolipids, which has important implications for biomarker development and therapeutic strategies.
In other contributions, Dr Clark's lab has participated (contribution of data and analysis) in several large-scale GWAS consortium studies that have led to the identification of PD genes, some of which are involved in the autophagy-lysosomal pathway. Her lab has also participated in a consortium study performing high-depth whole genome sequencing in control individuals to generate an Ashkenazi reference panel to support population-targeted personal genomics, performed the first genome wide association study and copy number variant study in a Parkinson's disease Ashkenazi Jewish population, and contributed to identification of a gene x gene interaction (PARK16 and LRRK2) in Parkinson's disease which involve two genes in the autophagy lysosomal pathway.
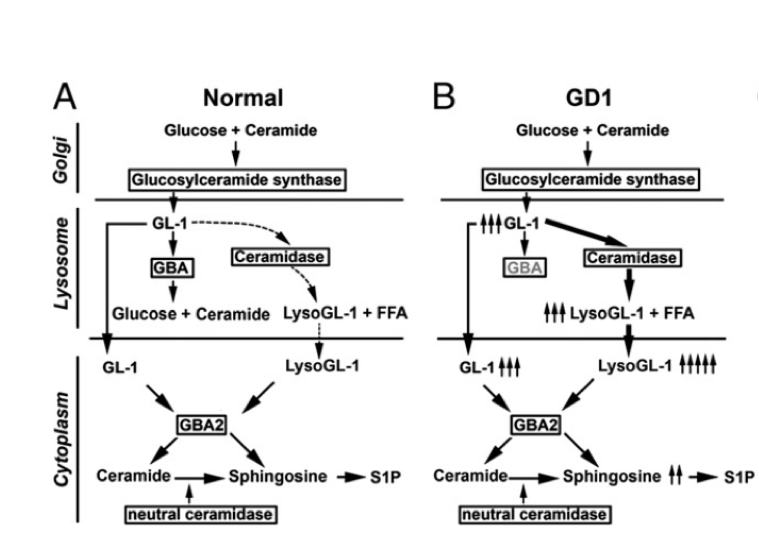 Figure Aberration of Sphingolipid pathway in GBA deficiency
|
 Figure Heat Maps showing significant changes in lipid classes. (PLOS ONE)
A) Heat map showing statistically significant changes in major lipid subclasses in LBD GBA mutation carriers compared to LBD wildtype, AD cases and controls and B) Heat map showing statistically significant changes in lipid classes in LBD GBA mutation carriers compared to LBD wildtype, AD cases and controls. The heat map columns reflect all significant lipid changes (q<0.05) in a diseased compared to control patients. The color bar represents the log2 value of the ratio of each lipid species. Statistical analysis for the AD and LBD Mutation samples was based on the one way analysis of variance followed by post hoc Fisher's least significant difference test while the LBD (wildtype) samples was based on Student’s T-test. A false discovery rate control was used to correct for multiple comparisons.
|
Lorraine Clark, PhD
Associate Professor of Pathology and Cell Biology (in the Taub Institute)
Lc654@cumc.columbia.edu
Joseph H. Lee, DrPH
Associate Professor of Epidemiology (in the Gerturde H. Sergievsky Center and Taub Institute)
Jhl2@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.