
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
June 2025
1: Early Proteasome Downregulation and Dysfunction Drive Proteostasis Failure in Alzheimer's Disease
2: HDAC Inhibitors Engage MITF and the Disease-Associated Microglia Signature to Enhance Amyloid β Uptake
3: The Role of Alpha-Synuclein in Synucleinopathy: Impact on Lipid Regulation at Mitochondria-ER Membranes
4: Associations Between Hearing Loss and Dementia in a Large Electronic Health Record System
2: HDAC Inhibitors Engage MITF and the Disease-Associated Microglia Signature to Enhance Amyloid β Uptake
3: The Role of Alpha-Synuclein in Synucleinopathy: Impact on Lipid Regulation at Mitochondria-ER Membranes
4: Associations Between Hearing Loss and Dementia in a Large Electronic Health Record System
Early Proteasome Downregulation and Dysfunction Drive Proteostasis Failure in Alzheimer's Disease
 |  |  | ||
| Shan Jiang, PhD | Malavika Srikanth, PhD | Natura Myeku, PhD |
In this study, recently published in BRAIN, we investigated a fundamental yet underexplored state of the ubiquitin–proteasome system (UPS) in Alzheimer’s disease (AD). We uncovered an early, intrinsic collapse in proteasome function that precedes overt protein tau aggregation, positioning the UPS as an early casualty and active contributor to proteostasis collapse in AD. Using human postmortem brain tissue, we combined proteasome activity assays and proteomics analyses to reveal that both 26S and 20S proteasomes are catalytically impaired in AD, even after purification, indicating structural deficits within the complexes themselves. Proteomic profiling revealed a marked depletion of core 19S and 20S subunits, as well as essential assembly chaperones, particularly in neuron-rich grey matter. Notably, 26S proteasomes from AD brains co-purified with aggregation-prone proteins like tau, α-synuclein, and p62/SQSTM1, suggesting that they are not only dysfunctional but also physically trapped within insoluble aggregates.
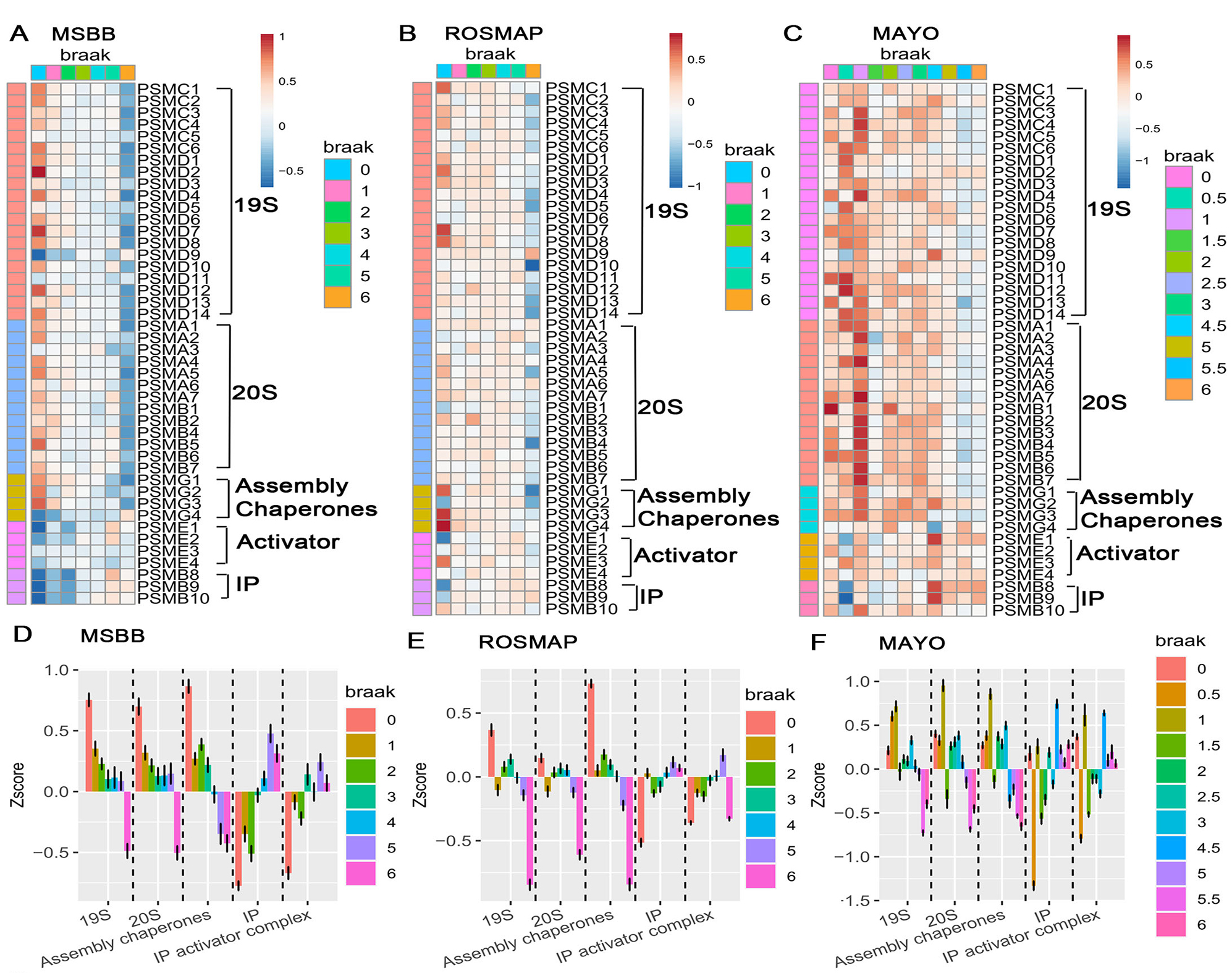
Figure 4. Progressive downregulation of constitutive proteasome subunits and differential responses of immunoproteasome across multiple AD cohorts. (A–C) Heatmaps showing the normalized expression (Z-scores) of constitutive proteasome subunits, assembly chaperones, and immunoproteasome (IP) components across increasing Braak stages for three independent cohorts of bulk-RNAseq datasets: (A) MSBB, (B) ROSMAP, and (C) Mayo Clinic Study of Aging. Each column represents a sample ordered by Braak stage (0-VI), while rows depict individual proteasome-related genes. Warmer colors (reds) indicate higher relative expression, and cooler colors (blues) indicate lower expression. A clear trend emerges where constitutive proteasome subunits (19S and 20S) and assembly chaperones decline with the advancing Braak stage, whereas immunoproteasome subunits show less consistent changes. (D–F) Boxplots of Z-scores for aggregated proteasome complexes and factors, 19S, 20S, assembly chaperones, immunoproteasome (IP), and IP activator complexes—grouped by Braak stage in (D) MSBB, (E) ROSMAP, and (F) Mayo cohorts. Each colored point or bar corresponds to a specific Braak stage. As Braak stage increases, 19S and 20S components consistently show a downward trend, while immunoproteasome and IP activator complexes remain relatively stable or exhibit compensatory changes.
The transcriptomics analyses of bulk RNA-seq datasets from three large AD cohorts (MSBB, ROSMAP, and Mayo) revealed a striking and progressive downregulation of constitutive proteasome subunit genes beginning at the earliest Braak stages, well before the appearance of tau pathology. These transcriptional deficits were further validated by snRNA-seq of two independent ROSMAP datasets, which revealed that this proteasome gene suppression is largely neuron-specific. In contrast, glial cells maintained or even upregulated proteasome-related transcripts, particularly those associated with the immunoproteasome, suggesting a cell–type–specific adaptive response to proteotoxic stress.
We then asked why neurons fail to mount a compensatory proteasome response. Under normal conditions of proteasome stress, cells activate the transcription factor NFE2L1 (Nrf1), which drives de novo synthesis of proteasome subunits through the “bounce-back” response. However, in AD brains, we observed that while Nrf1 transcript levels are elevated, its protein remains sequestered in the cytoplasm and fails to translocate to the nucleus, preventing effective activation of proteasome gene expression. We developed an “Nrf1 resist score” that quantifies this regulatory breakdown across disease stages, revealing that the disconnect between Nrf1 levels and proteasome gene expression intensifies as AD progresses. This nuclear translocation defect was further confirmed by subcellular fractionation and immunoblotting of human brain samples.
Our findings indicate that proteasome dysfunction is not merely a downstream consequence of AD pathology but may be a critical upstream event that promotes protein aggregation and neuronal vulnerability. Targeting proteasome function or restoring Nrf1 signaling represents a promising therapeutic strategy for preserving proteostasis in the aging brain.
Natura Myeku, PhD
Associate Professor of Pathology and Cell Biology
nm2631@cumc.columbia.edu
 |  | |
| Verena Haage, PhD | Philip L. De Jager, MD, PhD |
Disease-associated microglia (DAM) have been widely implicated in neurodegeneration, yet their precise role and regulatory mechanisms in the human brain remain incompletely defined. To address this, our team from the Taub Institute and the Center for Translational & Computational Neuroimmunology (CTCN), led by first author Dr. Verena Haage, in collaboration with colleagues from the Ludwig Center for Research on Neurodegeneration, developed a pharmacological approach to model human DAM in vitro—work recently published in Brain, Behavior, and Immunity.
Graphical Abstract
Using an in silico screening strategy, we prioritized two HDAC inhibitors, Vorinostat and Entinostat, which reproducibly engage key components of the human DAM transcriptional signature in microglia-like cells. These compounds not only induced expression of canonical DAM markers—including CD9, LPL, and SPP1—but also significantly upregulated MITF, a transcription factor recently proposed as a regulator of DAM-associated programs. Importantly, HDAC inhibitor-treated cells exhibited selective functional changes: enhanced uptake of amyloid β and dextran, reduced phagocytosis of bacterial particles, and attenuation of MCP-1 secretion in response to inflammatory stimuli. These findings were validated across two distinct human microglial model systems: the HMC3 microglia-like cell line and iPSC-derived microglia.
By providing a scalable and reproducible method to induce DAM-like states in human cells, our work advances the experimental toolkit for dissecting DAM biology. Moreover, our results support the emerging view that DAM signatures reflect at least two distinct transcriptional programs, underscoring the complexity of microglial polarization in neurodegenerative disease contexts. This platform offers new opportunities to mechanistically interrogate DAM function and to explore the translational potential of pharmacologically modulating microglial states.
Philip L. De Jager, MD, PhD
Weil-Granat Professor of Neurology (in The Taub Institute)
pld2115@cumc.columbia.edu
 |  |  | ||
| Peter Barbuti, PhD | Estela Area Gomez, PhD | Serge Przedborski, MD, PhD |
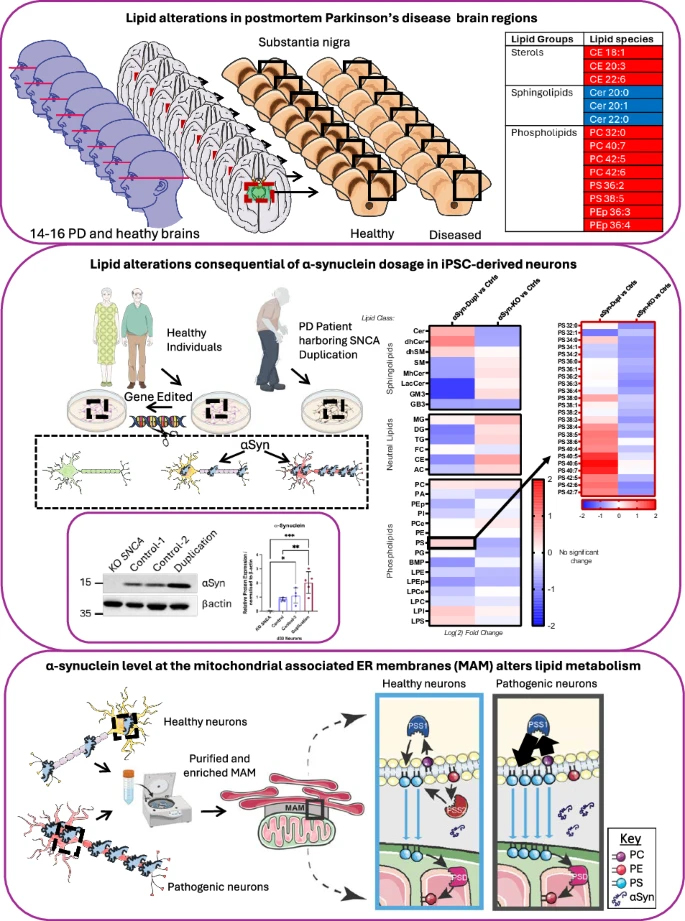
While alpha-synuclein (αSyn) dysfunction has long been recognized as central to the pathogenesis of synucleinopathies, the precise mechanisms linking αSyn to neuronal vulnerability remain incompletely understood. In our study, led by first author Dr. Peter Barbuti and conducted in collaboration with Dr. Estela Area-Gomez and colleagues from the Taub Institute and the Ludwig Center for Research on Neurodegeneration, we investigated the intersection of αSyn biology and lipid homeostasis, with a particular focus on mitochondria-associated ER membranes (MAMs), key subcellular domains regulating lipid metabolism.
Graphical Abstract
As reported in npj Parkinson’s Disease, through lipidomic profiling of postmortem brain samples from individuals with Parkinson’s disease (PD), multiple system atrophy (MSA), and healthy controls, we identified region- and disease-specific disruptions in sterols, phospholipids, and sphingolipids. The substantia nigra pars compacta (SNpc) of PD patients exhibited a distinct lipid signature, including elevated cholesteryl esters, reduced ceramides, and accumulation of polyunsaturated phosphatidylcholine and phosphatidylserine species—alterations only partially mirrored in MSA striatum. These findings suggest that αSyn-driven lipid dyshomeostasis may underlie selective regional vulnerability in PD.
To probe the mechanistic basis for these observations, we utilized human iPSC-derived neurons expressing varying levels of αSyn. These experiments revealed that αSyn localizes to MAM domains and regulates phosphatidylserine metabolism in a dosage-dependent manner. Notably, increased αSyn expression, as occurs with SNCA duplication, perturbs MAM lipid composition, disrupts sphingolipid and phospholipid homeostasis, and impairs PS decarboxylation—phenotypes consistent with lipid alterations observed in PD brain tissue. Together, these data implicate αSyn dosage and MAM dysfunction as key drivers of lipid dysregulation in synucleinopathy and highlight lipid metabolism as a promising axis for biomarker discovery and therapeutic intervention.
Serge Przedborski, MD, PhD
Page and William Black Professor of Neurology (in Pathology and Cell Biology and Neuroscience)
sp30@cumc.columbia.edu
Associations Between Hearing Loss and Dementia in a Large Electronic Health Record System
 |  | |
| James Noble, MD, MS | Justin Golub, MD, MS |
In the current study, led by Dr. Justin Golub (Columbia Otolaryngology–Head and Neck Surgery), we examined the association between audiometric hearing loss and dementia using a large, multi-institutional electronic health record (EHR) dataset. Leveraging data from nearly 32,000 adult patients who underwent formal audiometric testing at NYP/Columbia and Weill Cornell medical centers, we were able to assess hearing loss comprehensively through pure tone average, word recognition score, and speech reception threshold—three objective measures not consistently captured in national epidemiologic datasets.
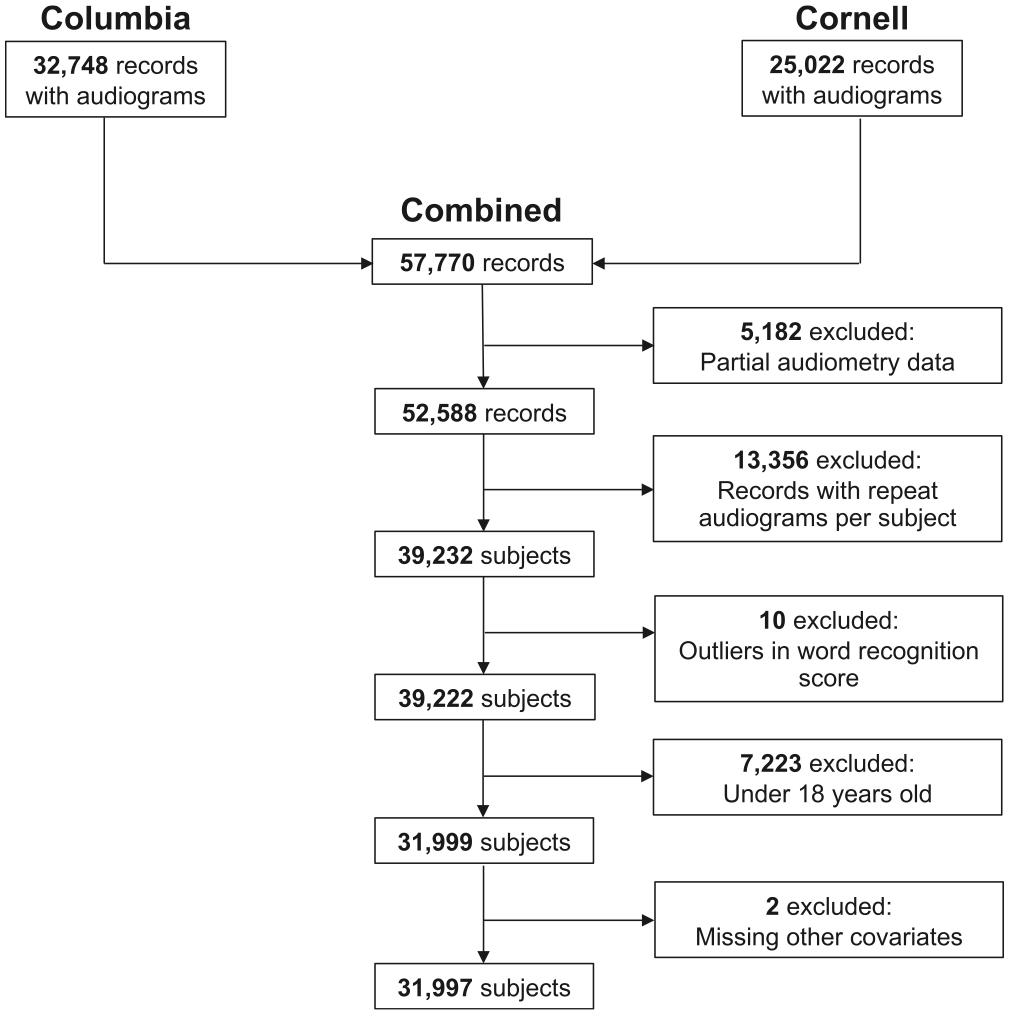
Figure 1: Flowchart of participant inclusion for analysis.
As recently reported in The Laryngoscope, our findings demonstrated that greater degrees of hearing loss, regardless of the metric used, were associated with significantly increased odds of dementia, even after controlling for key demographic and clinical confounders, including age, sex, cardiovascular risk, and study site. Notably, the relationship was consistent across different definitions of dementia, including diagnosis codes and medication use, underscoring the robustness of the association. The magnitude of risk observed—particularly among individuals with moderate to severe hearing loss—highlights the potential clinical significance of these findings.
While observational by design, this study reinforces prior work identifying hearing loss as a modifiable risk factor for cognitive decline and dementia. Furthermore, by demonstrating the feasibility and scientific value of leveraging large-scale EHR data, this work provides a framework for future longitudinal and interventional studies aimed at understanding the mechanistic pathways linking auditory and cognitive health, as well as evaluating the potential cognitive benefits of timely hearing loss treatment.
James Noble, MD, MS
Professor of Neurology (in the Taub Institute and the Gertrude H. Sergievsky Center)
jn2054@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.