
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
June 2018
1: » Excess Synaptojanin 1 Contributes to Place Cell Dysfunction and Memory Deficits in the Aging Hippocampus in Three Types of Alzheimer's Disease
2: » Preparation of Tau Oligomers After the Protein Extraction from Bacteria and Brain Cortices
2: » Preparation of Tau Oligomers After the Protein Extraction from Bacteria and Brain Cortices

Catherine Marquer, PhD
The three forms of AD—familial AD (FAD), Down syndrome-related AD (DS/AD), and sporadic AD (SAD)—share common clinical and neuropathology signatures, but their genetic causes differ. Early-onset FAD is caused by mutations in the APP, PSEN1, or PSEN2 gene and DS/AD is due to triplication of human chromosome 21 (Hsa21), while the most potent genetic risk factor for SAD is the ε4 allele of the APOE gene (APOE4). In the present study, published in Cell Reports, the laboratory of Dr. Catherine Marquer, along with collaborators from Taub and other institutions, asked whether elevated levels of synaptojanin 1 (SYNJ1), a key regulator of synaptic function, could be a common denominator to all three forms of AD.
Indeed, SYNJ1 levels are elevated in individuals at high risk for developing DS/AD (Martin et al., 2014) and SAD (Zhu et al., 2015), but what about FAD? Previous work performed in cellular and murine models in the Taub Institute by Drs. Gil Di Paolo and Tae Wan Kim supported that SYNJ1 may play a role in the physiology of FAD (Landman et al., 2006; McIntire et al., 2012), but its involvement in human FAD remained unknown. In the present study, Miranda, Herman, Cheng et al., in collaboration with Dr. Joseph H. Lee, asked whether SYNJ1 was associated with human FAD. They showed that variants in SYNJ1 are associated with age of onset and long-term memory deficits in an early-onset FAD cohort of Caribbean Hispanic families with the PSEN1-G206A founder mutation. Further, they demonstrated that variants in SYNJ1 are also associated with age of onset in the EFIGA cohort of late-onset FAD. These results in human FAD cohorts thus strongly suggest that SYNJ1 may play a role in all three forms of AD.
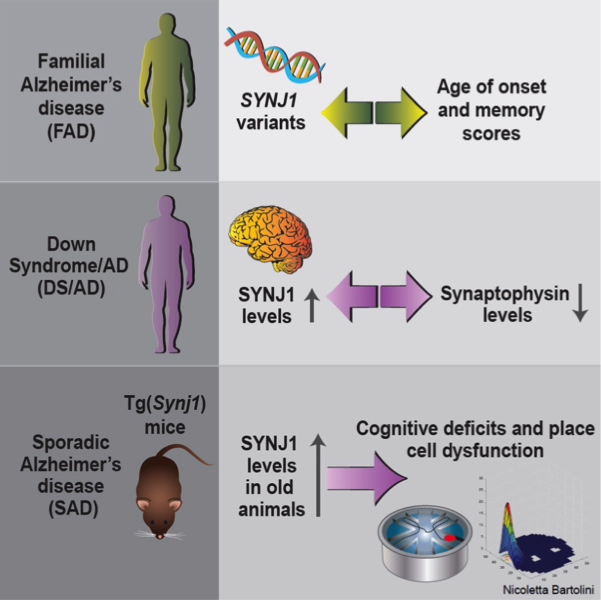
Figure: Findings from the Marquer lab, combining human genetics, human autopsy brain samples, and behavior and in vivo electrophysiology studies in a transgenic mouse model, strongly support that SYNJ1 plays a role in memory deficits in all three forms of AD
Dr. Marquer and co-investigators then explored whether the elevated levels of SYNJ1 observed in DS/AD and SAD directly affected cognition. For DS/AD, they teamed up with Drs. Elizabeth Head, Frederick A. Schmitt and Ira T. Lott (University of Kentucky and University of California, Irvine). They found that increased SYNJ1 levels in autopsy brains from adults with DS/AD are inversely correlated with synaptophysin levels, which are commonly used as an estimation of the number of synapses. No such correlation was observed in younger individuals with DS, whose SYNJ1 levels are similar to those of disomic controls.
For SAD, they recapitulated the SYNJ1 overexpression levels described in SAD in a transgenic mouse model, Tg(Synj1) (Voronov et al., 2008). Their behavior experiments revealed that, while 9-month old transgenic animals performed similarly to the controls, 19-month old transgenic animals showed hippocampal cognitive deficits. Marquer and colleagues found that increased levels of Synj1 did not impair learning per se in older animals, but caused a specific defect in long-term memory retention. Using in vivo recordings, in collaboration with Dr. Abid Hussaini, they showed that this defect is due to hippocampal hyperexcitability and, more specifically, to a dramatic alteration in the spatial reproducibility of hippocampal place fields, which may affect memory consolidation.
Taken together, their data strongly argue that SYNJ1 plays a role in the function of place cells in the aging hippocampus, with critical implications for memory deficits in all three forms of AD and their possible treatment.
Catherine Marquer, PhD
Assistant Professor of Pathology and Cell Biology (in the Taub Institute)
cm3244@cumc.columbia.edu
Preparation of Tau Oligomers After the Protein Extraction from Bacteria and Brain Cortices

Ottavio Arancio, MD, PhD
Tau protein is the main component of neurofibrillary tangles that constitute one of the histological markers of Alzheimer’s disease (AD). Initially, it was thought that neurofibrillary tangles are the major toxic entities in AD, although recent findings seem to challenge this idea, suggesting that synaptic dysfunction and neuronal dearth are mainly related to early-stage tau oligomers. There is a growing body of evidence suggesting that tau oligomers induce the early neuronal damage and, subsequently, the learning and memory impairment characterizing AD. Injection of the full-length recombinant oligomerized tau in the mouse brain activates the mitochondrial apoptotic pathway and reduces the levels of proteins participating in synaptic transmission. Accordingly, tau oligomers, not fibrils or monomers, impair memory consolidation in mice injected with tau oligomers purified from human Alzheimer’s brains or recombinant human tau. In particular, tau oligomers disrupt storage of new memories rendering mice incapable of remembering new information. Interestingly, tau oligomers are responsible for the neuronal damage occurring in AD, even before the aggregation of Αβ fibrils.
Published recently in Methods in Molecular Biology, the Taub Institute laboratory of Dr. Ottavio Arancio conducted a study aiming to establish an efficient and cost effective method for isolating tau protein from bacterial cells and cortices of AD patients or mice. After the final step of purification, isolated soluble tau was oligomerized, achieved via introduction of disulfide bonds. Even though the species that induced the toxicity could not be identified, Dr. Arancio’s protocol for producing tau oligomers proved to efficiently affect key features of Alzheimer’s disease, including synaptic plasticity and memory.
Ottavio Arancio, MD, PhD
Professor of Pathology and Cell Biology (in the Taub Institute)
oa1@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.