
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
May 2025
1: CLU Alleviates Alzheimer's Disease-Relevant Processes by Modulating Astrocyte Reactivity and Microglia-Dependent Synaptic Density
2: Selective Removal of Astrocytic PERK Protects Against Glymphatic Impairment and Decreases Toxic Aggregation of β-Amyloid and Tau
3: APOE ε4-Associated Heterogeneity of Neuroimaging Biomarkers Across the Alzheimer's Disease Continuum
2: Selective Removal of Astrocytic PERK Protects Against Glymphatic Impairment and Decreases Toxic Aggregation of β-Amyloid and Tau
3: APOE ε4-Associated Heterogeneity of Neuroimaging Biomarkers Across the Alzheimer's Disease Continuum
 |  |  | ||
| Philip L. De Jager, MD, PhD | Vilas Menon, PhD | Tracy Young-Pearse, PhD (Harvard) |
The gene Clusterin (CLU) has been implicated in Alzheimer’s disease (AD) through genome-wide association studies, but its precise molecular role in the disease has remained elusive. In a collaborative study led by Tracy Young-Pearse’s group at Brigham and Women’s Hospital, we investigated the functional consequences of reduced CLU expression in astrocytes. Leveraging our large-scale single-nucleus RNA-seq dataset (Green et al., Nature 2024), we observed significantly lower CLU expression in astrocytes from post-mortem brain tissue of individuals with AD.
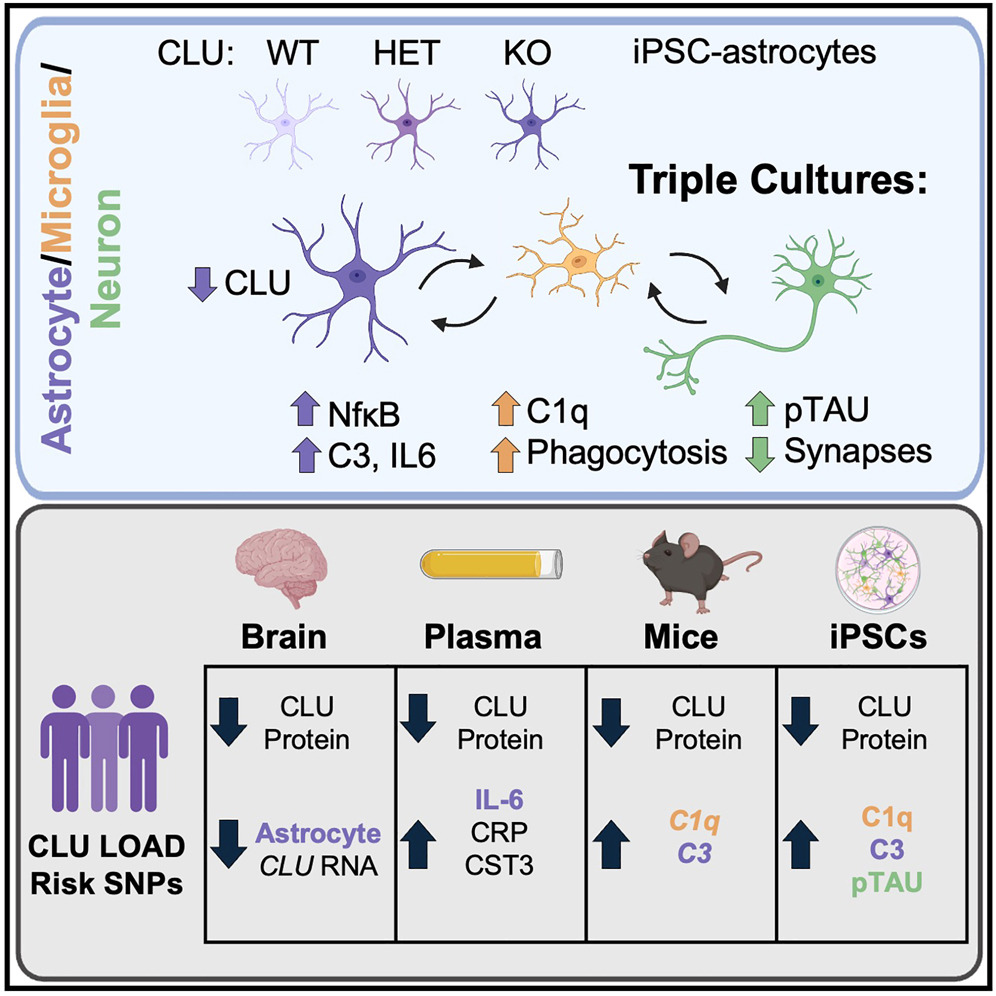
Graphical abstract.
Building on this, the Young-Pearse lab generated CLU-deficient astrocytes from induced pluripotent stem cells and conducted unbiased proteomic analyses. As recently reported in Neuron, this revealed heightened nuclear factor κB (NF-κB)-dependent signaling and increased secretion of complement C3—both indicators of an amplified inflammatory state. Functionally, these reactive astrocytes triggered microglia-dependent changes, including elevated extracellular apolipoprotein E (APOE), increased phosphorylated tau, enhanced microglial phagocytosis, and reduced synapse numbers.
By integrating mouse and human cellular models with deep profiling of human plasma and brain tissue, the study further showed that individuals carrying CLU AD-risk alleles exhibit both reduced CLU protein levels and elevated inflammatory markers. Together, these findings establish a mechanistic link between genetic risk, astrocyte reactivity, microglial behavior, and synaptic vulnerability. The data support a model in which CLU upregulation acts as a protective response to neuropathology, while diminished CLU levels in astrocytes increase susceptibility to neurodegeneration.
Vilas Menon, PhD
Assistant Professor of Neurological Sciences (in Neurology and the Taub Institute)
vm2545@cumc.columbia.edu
Philip L. De Jager, MD, PhD
Weil-Granat Professor of Neurology(in The Taub Institute)
pld2115@cumc.columbia.edu
 |  | |
| Kai Chen, PhD | Tal Nuriel, PhD | |

|  | |
| Osama Al Dalahmah, MD, PhD | Guang Yang, PhD |
The glymphatic system, often described as the brain’s “waste disposal network”, plays an important role in clearing potentially harmful proteins such as β-amyloid (Aβ) and hyperphosphorylated tau, both key hallmarks of Alzheimer’s disease (AD). When this system becomes impaired, these toxic proteins can accumulate, contributing to disease progression. In a recent study, conducted in collaboration with Drs. Tal Nuriel and Osama Al-Dalahmah of the Taub Institute, we explored the molecular mechanisms underlying glymphatic dysfunction in AD. Using postmortem human brain tissue and two transgenic mouse models (5XFAD and PS19), our research uncovered an astrocyte signaling pathway that could be targeted to protect glymphatic function and reduce the buildup of toxic proteins.

Figure 1: Schematic summary. In AD mouse models, chronic activation of astrocyte PERK-eIF2α signaling suppresses protein synthesis and disrupts perivascular AQP4 localization via CK2-dependent mechanisms. Inhibiting astrocytic PERK restores glymphatic function, reduces AD-related pathology, and improves cognitive performance.
As recently reported in Neuron, we found that the PERK-eIF2α signaling pathway, a key branch of the unfolded protein response activated during cellular stress, is chronically overactive in astrocytes in AD brains. This sustained activation suppresses protein synthesis and disrupts the perivascular positioning of aquaporin-4 (AQP4), an astrocyte water channel essential for glymphatic flow, via casein kinase 2 (CK2)-dependent mechanisms. Importantly, our results showed that both genetic deletion and pharmacological inhibition of PERK in astrocytes restored AQP4 localization, enhanced glymphatic clearance, reduced Aβ and tau pathology, and improved cognitive performance in mouse models. These findings underscore the critical role of the astrocyte PERK-CK2-AQP4 axis in glymphatic dysfunction and AD progression, positioning this pathway as a promising therapeutic target for future treatments.
Kai Chen, PhD
Associate Research Scientist in the Department of Anesthesiology
kc3378@cumc.columbia.edu
Guang Yang, PhD
Associate Professor in the Department of Anesthesiology
gy2268@cumc.columbia.edu
APOE4-Associated Heterogeneity of Neuroimaging Biomarkers Across the Alzheimer's Disease Continuum
 | | | ||
| Jason Mares | Vilas Menon, PhD | Tal Nuriel, PhD |
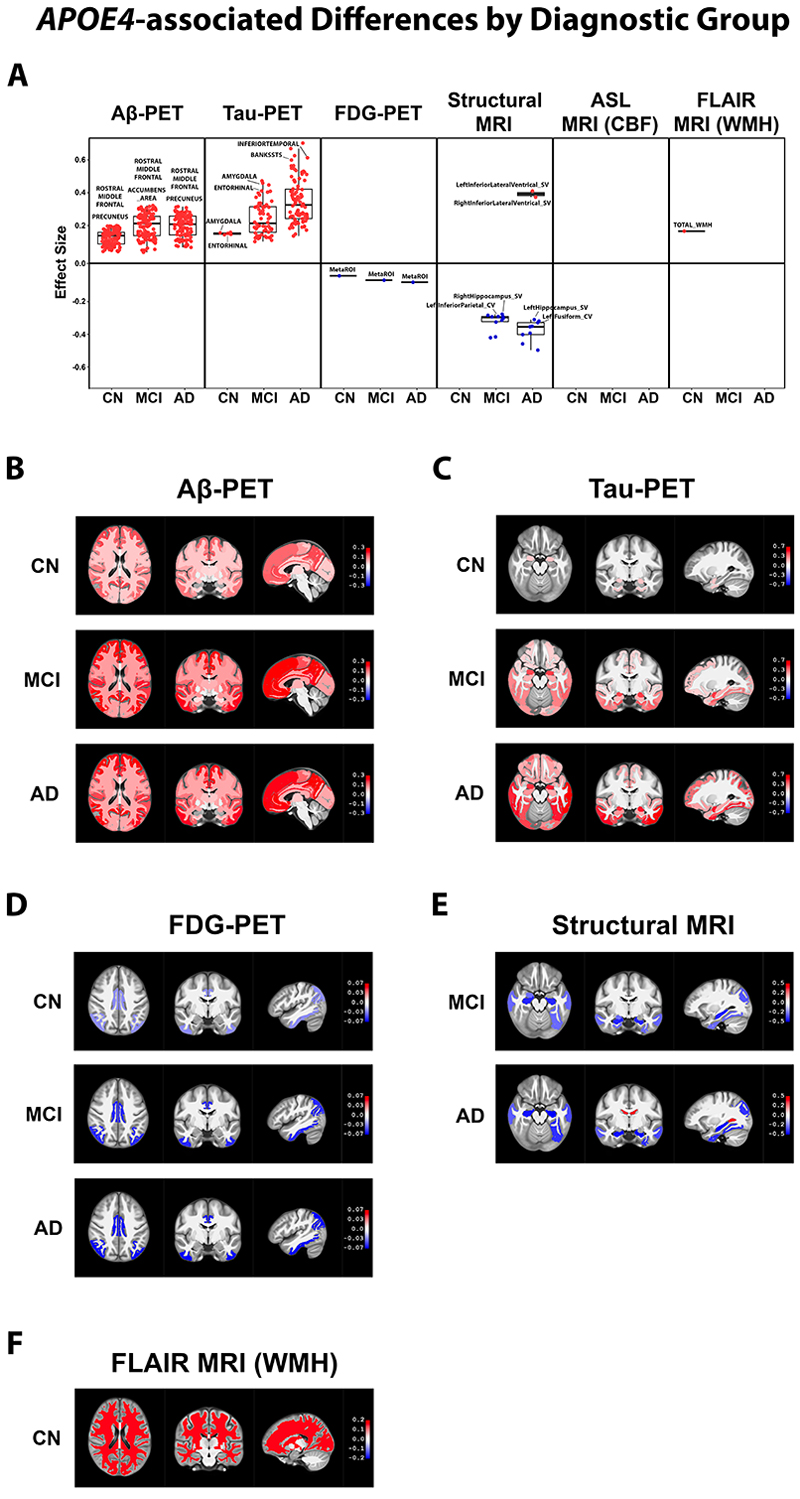
Figure 1. APOE4–associated differences by diagnostic group. We performed linear mixed‐effects analyses comparing regional neuroimaging biomarker levels between APOE4 carriers versus non‐carriers, classified into one of three diagnostic groups: CN, MCI, or AD participants. Regions with significantly different biomarker levels (p ‹ 0.05) between the APOE4 carriers and non‐carriers in each diagnostic group were graphed onto box plots with two of the top regions labeled (A). We also rendered the regions onto a human brain template (displayed axially, coronally, and sagittally) using different shades of red (upregulated in APOE4 carriers) or blue (downregulated in APOE4 carriers) that corresponds to the magnitude of their statistical difference (beta) between APOE4 carrier groups (B)–(F). We show the rendered biomarker differences from each analysis that resulted in at least one statistically different region, which in this diagnostic group–level comparison included Aβ PET results (B), tau PET results (C), FDG PET results (D), structural MRI results (E), and FLAIR MRI WMH results (F). Aβ, amyloid beta; AD, Alzheimer's disease; APOE, apolipoprotein E; ASL, arterial spin labeling; CBF, cerebral blood flow; CN, cognitively normal; FDG, fluorodeoxyglucose; FLAIR, fluid‐attenuated inversion recovery; MCI, mild cognitive impairment; MRI, magnetic resonance imaging; PET, positron emission tomography; WMH, white matter hyperintensity
Apoliprotein E ε4 (APOE4) is the primary genetic risk factor for the late-onset, sporadic form of Alzheimer’s disease (AD). As such, researchers have extensively investigated the reasons why APOE4 increases an individual’s susceptibility to AD. However, much less research has been conducted to identify the differences in disease presentation that occur between APOE4 carriers vs. non-carriers. To help address this topic, we performed a comprehensive analysis of neuroimaging biomarkers in the brains of APOE4 carriers vs. non-carriers who participated in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Specifically, we processed ADNI data from six neuroimaging biomarkers: florbetapir positron emission tomography (PET) measurements of amyloid beta (Aβ) deposition, flortaucipir PET measurements of tau accumulation, fluorodeoxyglucose (FDG) PET measurements of glucose uptake, structural magnetic resonance imaging (MRI) measurements of brain volume, arterial spin labeling (ASL) MRI measurements of cerebral blood flow (CBF), and fluid‐attenuated inversion recovery (FLAIR) MRI measurements of white matter hyperintensities (WMHs). Furthermore, we compared the regional levels of each of these biomarkers in APOE4 carriers vs. non-carriers at three separate ADNI-determined diagnostic stages: cognitively normal (CN), mild cognitive impairment (MCI), and AD. The analysis was conducted under my supervision and primarily performed by Jason Mares, a highly skilled bioinformatician in Dr. Vilas Menon’s group.
Overall, we observed extensive heterogeneity in how these neuroimaging biomarkers present in the brains of APOE4 carriers vs. non-carriers, as reported in Alzheimer’s & Dementia. In particular, we observed robust APOE4–associated increases in Aβ deposition throughout the brain, in all three diagnostic groups. We also observed APOE4–associated increases in tau pathology, decreases in glucose uptake, and increases in brain atrophy, which expanded in regional scope and magnitude with disease progression. Significant sex‐ and age‐related differences in APOE4–associated neuroimaging biomarker heterogeneity were also observed, with female APOE4 carriers displaying notable increases in pathological presentation for a variety of biomarkers. Lastly, specific regional differences in Aβ deposition, tau accumulation, glucose uptake, ventricle size, and WMHs were observed in CN participants who converted to MCI/AD during their participation in ADNI, which we showed may hold potential predictive value for determining the likelihood of MCI/AD conversion among cognitively unimpaired individuals.
Our study highlights the need for genotype-specific approaches in AD diagnostics and interventions, given the clear biological heterogeneity associated with an individual’s APOE4 status.
Tal Nuriel, PhD
Assistant Professor of Pathology and Cell Biology (in the Taub Institute) at CUMC
tn2283@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.