
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
May 2022
1: FMNL2 Regulates Gliovascular Interactions and Is Associated with Vascular Risk Factors and Cerebrovascular Pathology in Alzheimer’s Disease
2: Molecular Insights into Cell Type-Specific Roles in Alzheimer's Disease: Human Induced Pluripotent Stem Cell-Based Disease Modeling
3: Effects of Eph/Ephrin Signalling and Human Alzheimer's Disease-Associated EphA1 on Drosophila Behaviour and Neurophysiology
2: Molecular Insights into Cell Type-Specific Roles in Alzheimer's Disease: Human Induced Pluripotent Stem Cell-Based Disease Modeling
3: Effects of Eph/Ephrin Signalling and Human Alzheimer's Disease-Associated EphA1 on Drosophila Behaviour and Neurophysiology
 |
 |  | |
| Caghan Kizil, PhD | Badri N. Vardarajan, PhD, MS | Richard Mayeux, MD, MSc |
Cardiovascular and cerebrovascular diseases co-exist in most Alzheimer’s disease (AD) patients, and cardio/cerebrovascular risk factors (CVRFs) are associated with the risk of AD. However, the genetic variation related to the risk factors and the molecular mechanisms underlying the interplay of CVRF and genetics in AD are not well understood. In our publication in Acta Neuropathologica, we analyzed large multiethnic Alzheimer’s disease cohorts to understand the role of cardiovascular risk in AD. We created a CVRF score comprised of hypertension, diabetes, heart disease and body-mass-index and tested the interaction of the score with genetic variants in conferring risk of AD. We found that genetic alterations in one gene, FMNL2, interact with enhanced risk score for CVRFs, to modify the risk of developing AD. Analyses of transcriptomics data from the frontal cortex in human brains showed that FMNL2 expression increased in patients that have pathological hallmarks of AD, as well as chronic infarcts. We performed a causal mediation analysis and found that both amyloid and tau pathology increased FMNL2 expression which, in turn, resulted in higher risk of AD, suggesting that AD brains enhance the activity of this gene.
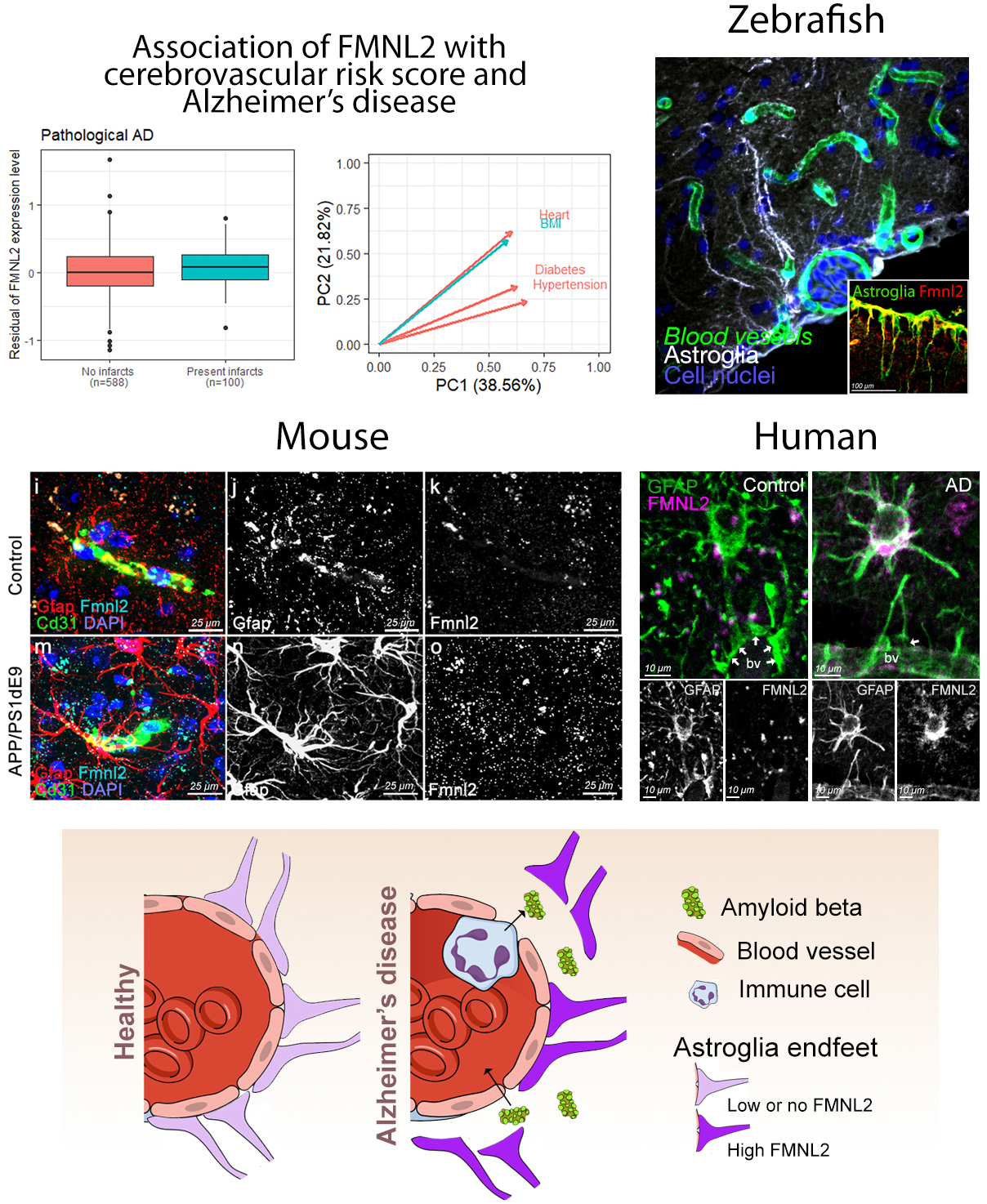
Figure: (A) Gene association and causal mediation analyses showed FMNL2 expression in the brain increases with Alzheimer's disease (AD) pathology in presence of cardiovascular risk factors (CVRFs). (B) Zebrafish model showed that gliovascular (GV) interactions can relax upon AD pathology to enhance clearance and immune activity. These are impaired by loss of FMNL2 function. GV relaxation through FMNL2 activity is evolutionarily conserved as shown in chronic AD model in mice (C) and in post-mortem human brains (D). (E) FMNL2 activity is required for the pathology-induced GV remodeling, activity of immune cells and clearance. Genetic variants of FMNL2 and CVRFs alter this process and modify the risk of developing AD.
FMNL2 belongs to the formin family of proteins, which are implicated in AD pathology and involved in regulating cytoskeleton, cellular morphology, and contact. To analyze the functional involvement of FMNL2 in AD pathology, we used an established acute model of amyloidosis in adult zebrafish brain. We found that FMNL2 (Fmnl2a in zebrafish) is mainly present in astroglial cells and neurons, and its expression is enhanced upon acute application of human amyloid beta42. The astroglia that express Fmnl2a are establishing a close contact with the blood vessels. After amyloid pathology, these cells retract their endfeet on the blood vessels, generate a larger perivascular space, and augment the permeability of the blood-brain-barrier. Interestingly, when Fmnl2 function was blocked, amyloid pathology-induced endfeet retraction was severely impaired, the abundance of active microglial cells in the brain were reduced, and amyloid aggregation around the blood vessels increased. Therefore, we hypothesized that enhanced FMNL2 activity after amyloid pathology is a response to cope with amyloid aggregations.
To validate whether FMNL2 upregulation in AD is consistent and evolutionarily conserved in chronic models of amyloidosis in mammals, we used an AD mouse model and showed that Fmnl2 increased around the blood vessels concomitant to the glial endfeet retraction. This phenomenon was not related to the generic gliotic response, as amyloid toxicity but not traumatic injuries led to enhanced Fmnl2. Finally, we showed that in post-mortem human brains with AD and primary age-related tauopathy, FMNL2 expression was also enhanced at gliovascular junctions with AD and cerebrovascular pathology.
Our findings in humans, mouse and zebrafish showed that FMNL2 is minimally expressed at the blood vessel and glial junctions in a healthy state of the brain, but its levels were elevated upon CVRFs and AD. Therefore, we propose FMNL2 as a critical disease-related player in vascular niche of the brain. FMNL2 could determine the brain’s ability to cope with amyloid and tau pathology by controlling toxic protein clearance, immune system activity, and regulation of blood-brain-barrier dynamics. We consider that FMNL2 is part of a larger set of genetic and molecular network that underlies the interaction between CVRF and AD. Genetic variants of FMNL2 could reduce the protein function and, therefore, lead to exacerbated disease progression through impaired protein clearance and microglial activation. Our study, which is also featured on the CUIMC Newsroom, is an example of how clinical studies and basic science with animal models could be harmonized to discover new disease mechanisms, which could open new clinical directions and drug development efforts.
Richard Mayeux, MD, MSc
Gertrude H. Sergievsky Professor of Neurology, Psychiatry and Epidemiology (in the Taub Institute and the Gertrude H. Sergievsky Center)
rpm2@cumc.columbia.edu
Badri N. Vardarajan, PhD, MS
Assistant Professor of Neurological Science (in Neurology, the Gertrude H. Sergievsky Center, and the Taub Institute)
bnv2103@cumc.columbia.edu
Caghan Kizil, PhD
Visiting Professor of Neurological Sciences (in Neurology)
ck2893@cumc.columbia.edu
 |  | |
| Wenhui (Wendy) Qu, PhD | Gunnar Hargus, MD, PhD |
Alzheimer’s disease (AD) is characterized by progressive loss of cognitive function and accounts for at least half of patients with dementia. As a currently incurable disease, AD places a severe burden on patients and families as well as healthcare systems as society ages. Taking advantage of human genetic studies, rodent models carrying AD mutations successfully mimic the age-related neurodegenerative aspect of AD and they have provided invaluable molecular insights into cell type-specific mechanisms of AD pathogenesis. However, attempts to translate these insights from rodent studies to clinical trials in AD patients have failed, highlighting the need for better models. The expression and cellular regulation of several key AD-related proteins differ significantly between humans and rodents, which may have contributed to negative outcomes. Emerging evidence has suggested the importance of utilizing human cells, such as induced pluripotent stem cell (iPSC)-derived brain cells, to model human neurodegenerative diseases.
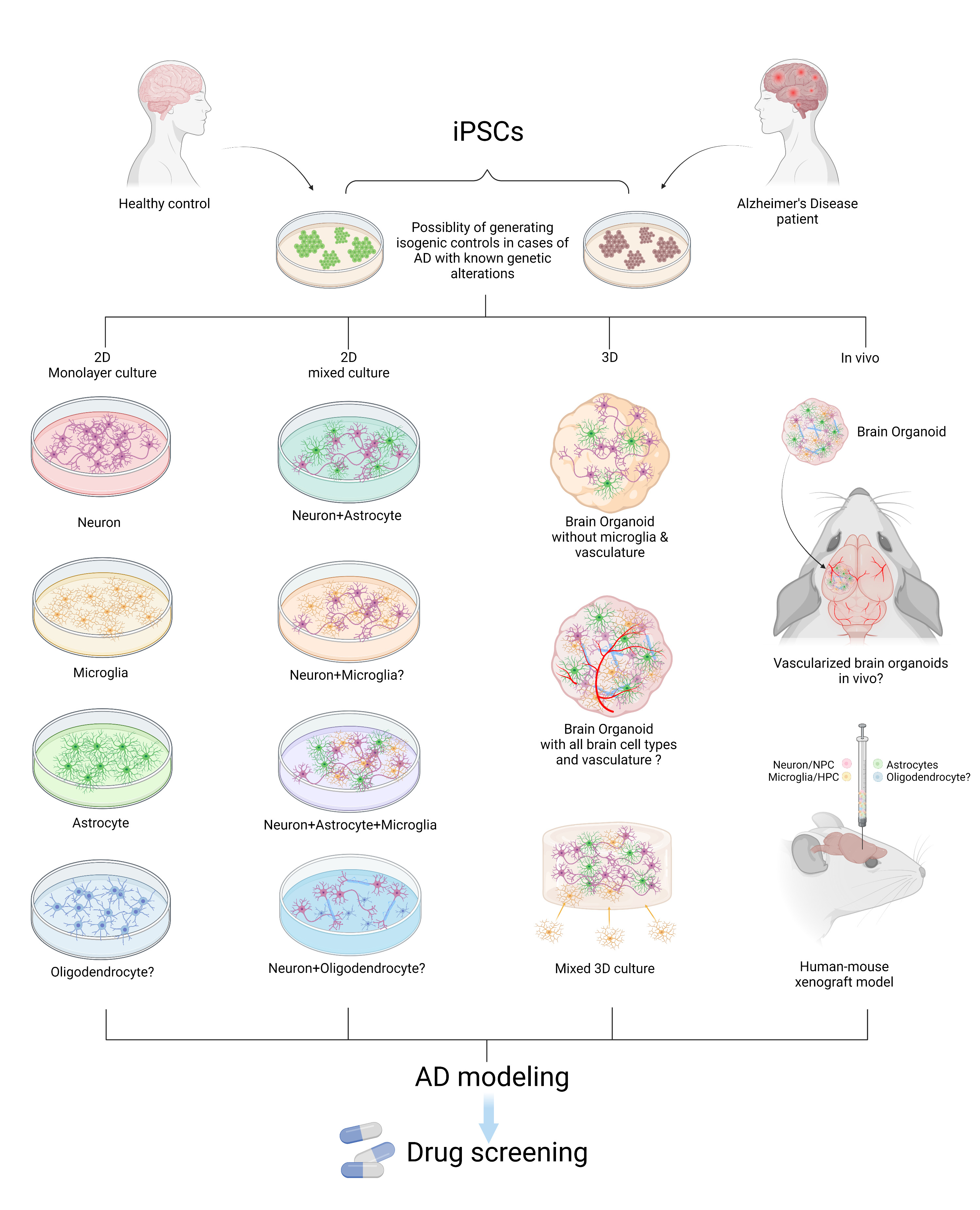
Figure 1. Modeling AD with patient iPSCs. AD patient or healthy control-derived iPSCs can be differentiated into different brain cell types including neurons, microglia, astrocytes, and oligodendrocytes. In vitro and in vivo AD modeling strategies can be utilized to model AD and they can be applied for drug screening. “?” indicates available technology yet to be used in AD research.
In this review, recently published in Neuroscience, we summarize reported studies on human iPSC models of AD (Figure 1). We first provide an overview of the function of different major brain cell types in the pathogenesis of AD, including neurons, microglia, and astrocytes. We then review how AD patient derived iPSCs, carrying familial (including APP, PSEN1, and PSEN2) or sporadic (e.g. APOE4, TREM2) AD mutations/variants, have been utilized to study disease phenotypes in these cell types. For example, several reports demonstrate Aβ and tau pathology, synaptic dysfunction and increased stress responses in AD iPSC-derived neurons, as well as altered expression of pro-inflammatory mediators in AD iPSC-derived astrocytes and microglia (Figure 2). We also demonstrate how patient iPSCs models have been used to study both cell-autonomous and non-cell-autonomous mechanisms of disease development in AD. In addition to pure or mixed cell 2D-monolayer cultures, 3D-cultures and brain organoids as well as human iPSC cell-transplantation mouse models have been applied to study cell-intrinsic phenotypes or cell-cell interaction in vitro and in vivo (Figure 1).
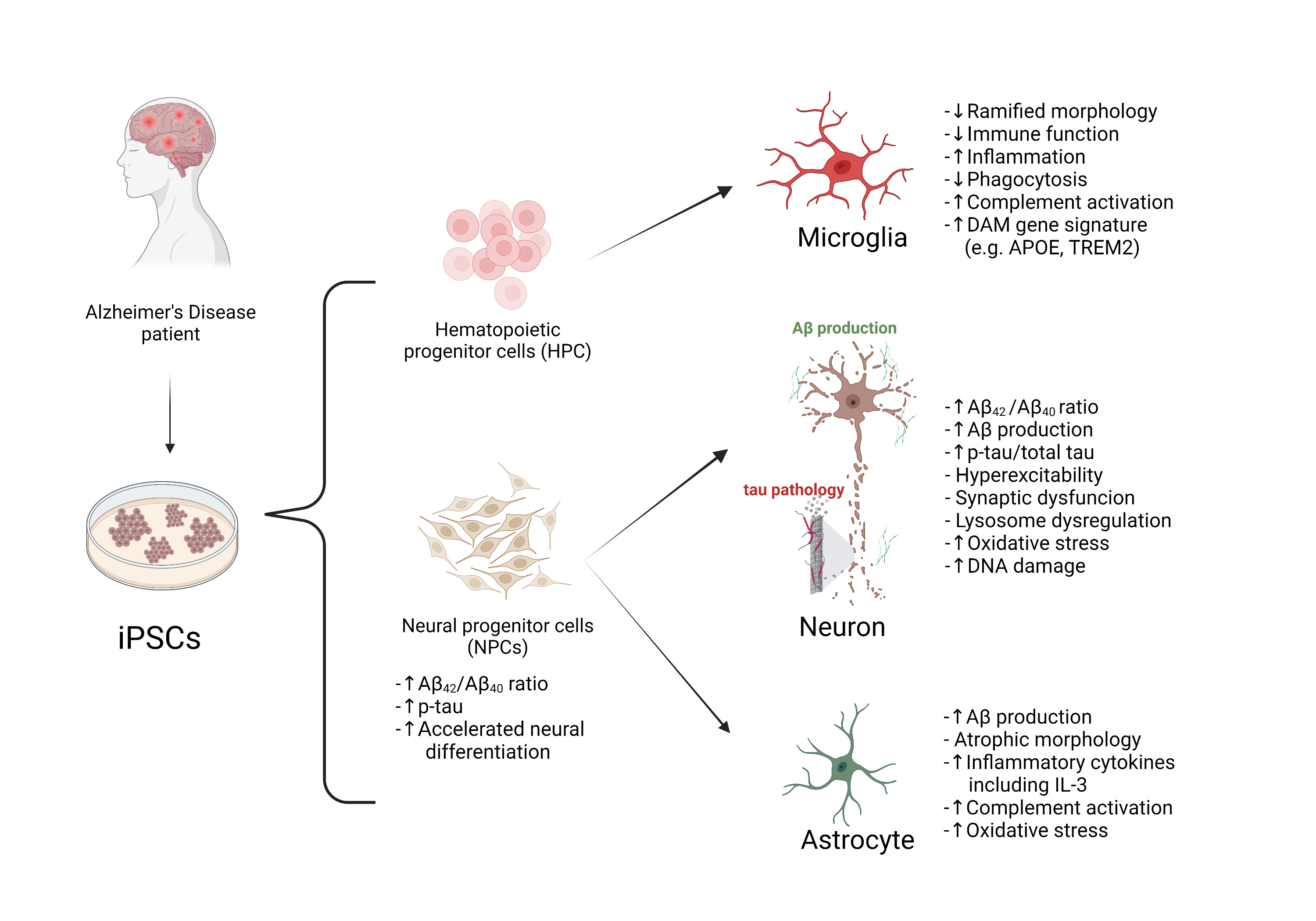
Figure 2. Summary of disease phenotypes in AD iPSC-derived brain cells. AD-related phenotypes have been described mainly in neurons, microglia, NPCs, and astrocytes.
While we discuss limitations and areas of improvement for using iPSCs for disease modeling, we also highlight the strong potential of iPSC technology for biomedical research on neurodegenerative diseases such as AD. For instance, the iPSC disease modeling technology can be applied to drug screening in vitro and in vivo, and, potentially, for the development of patient-specific treatment strategies. These models can also be used to validate key findings in rodent studies and to further explore many unsettled questions including the function of AD risk factors in different brain cell types. Combining iPSC technology with genetic and molecular studies in human AD patients and animal models should lead to important advancements in both the understanding and treatment of this devastating disease.
Wenhui (Wendy) Qu, PhD
Postdoctoral Research Scientist in the Department of Pathology and Cell Biology (in the Taub Institute)
wq2163@cumc.columbia.edu
Gunnar Hargus, MD, PhD
Assistant Professor of Pathology and Cell Biology (in the Taub Institute)
gh2374@cumc.columbia.edu
 |
 |  | |
| Roger Lefort, PhD | Ismael Santa-Maria Perez, PhD | Yoon A. Kim, PharmD, PhD |
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease, placing a great burden on patients, their caregivers and society. Characterization of the genetic landscape of AD and related dementias provides a unique opportunity for a better understanding of the associated pathophysiological processes, potentially leading to a future cure. Genome-wide association studies (GWAS) have identified several genes associated with late onset Alzheimer's disease (LOAD) including common variants (Carrasquillo et al., 2011; Hollingworth et al., 2011; Naj et al., 2011), and from our Taub colleagues Drs. Mayeux and Vardarajan, a nonsynonymous proline to leucine substitution at the highly conserved protein coding position 460 (P460L) of the receptor tyrosine kinase EphA1 in Caribbean Hispanic families and Caucasians (nominally significant; Vardarajan et al., 2015). However, demonstration of whether and how these genetic variants cause pathology is largely lacking. In a recent article published in Neurobiology of Disease, we set out to characterize the effects of AD-associated EphA1 and investigate the role of Eph/ephrin signalling on disease relevant behavior and physiology using a Drosophila model.
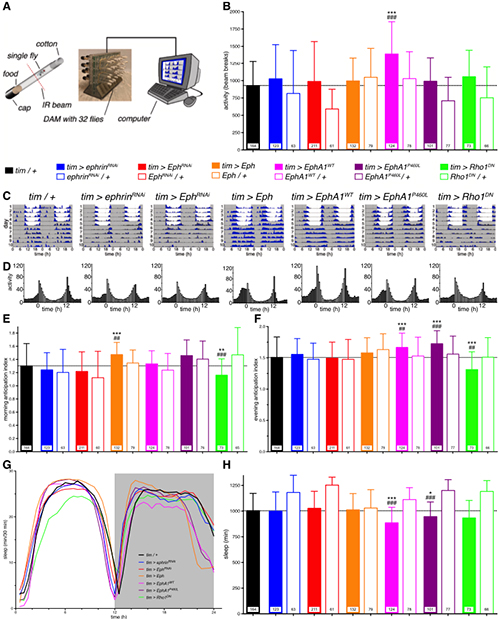
Figure 3. Eph/ephrin signalling disrupts locomotor activity and sleep.
(A) Diagram of the automated fly tracking setup. Individual flies are placed in food containing tubes and loaded into a DAM monitor, where an infrared beam intersects the tube, recording when the fly travels along the tube breaking the beam. The DAM monitor is connected to a computer recording the accumulated number of beam breaks. (B) Total activity levels, measured as the average daily number of beam crosses showed an increase in activity of flies mis-expressing the human wild-type receptor throughout the circadian clock (tim > EphA1WT). Genotypes colour coded as indicated. (C) Double-plotted actograms of exemplary flies depicting the daily activity levels of control (tim > +) and experimental flies (as indicated). Flies were recorded for the first 5 days in a 12 h:12 h LD cycle (grey, lights off; white, lights on) and then released in constant dark conditions for 5 days (DD). (D) Histograms showing the 5-day average activity levels of all flies in LD for the same genotypes. (E) Morning and (F) evening anticipation indices demonstrating an increased morning anticipation for fly Eph over-expressing flies and increased evening anticipation for EphA1WT and EphA1P460L flies while decreased anticipation for Rho1DN flies. (G) Sleep distribution profiles, plotted as time asleep within 30 min bins, over the day (grey, lights off; white, lights on) and (H) total daily sleep for the LD condition showed reduced sleep for flies mis-expressing either human EphA1 receptor. Genotypes colour coded as indicated. Bars, means; whiskers, SD; n, numbers in bars; * p < 0.05, **, ## p < 0.01, ***, ### p < 0.001; Kruskal-Wallis with Dunn's post hoc test compared to Gal4 (*) and to respective UAS controls (#).
Working in collaboration with Drs. Hodge and Buhl (University of Bristol), we first generated a Drosophila model expressing human wild-type and P460L mutant EphA1, with the aim of testing the effects of Eph (receptor)/ephrin (ligand) signalling on AD-relevant behavior and neurophysiology. We found flies expressing the wild-type or mutant EphA1 receptor were hyper-aroused, presenting reduced sleep, a stronger circadian rhythm, increased clock neuron activity and excitability. Further, we investigated whether over-expression of endogenous fly Eph receptor and RNAi-mediated knock-down of Eph and its ligand ephrin affected sleep architecture and neurophysiology. We found fly Eph over-expression led to stronger circadian morning anticipation while ephrin knock-down impaired memory. A dominant negative form of the GTPase Rho1, a potential intracellular effector of Eph, led to hyper-aroused flies, memory impairment, less anticipatory behavior, and neurophysiological changes. Altogether, these novel results demonstrate a role of Eph/ephrin signalling in a range of behaviors similarly affected in AD patients. This study sets a starting point for future investigations uncovering novel mechanisms of AD opening new therapeutic interventions.
Roger Lefort, PhD
Assistant Director for Research Integrity and Compliance
rl2040@cumc.columbia.edu
Ismael Santa-Maria Perez, PhD
Assistant Professor of Pathology and Cell Biology (in the Taub Institute)
is2395@cumc.columbia.edu
Yoon A. Kim, PharmD, PhD
Postdoctoral Research Scientist in Pathology and Cell Biology (in the Taub Institute)
yk2477@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.