Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
November 2016
#1 Activating Transcription Factor 4 (ATF4) Modulates Rho GTPase Levels and Function via Regulation of RhoGDIα
#2 Brain Regions Involved in Arousal and Reward Processing are Associated with Apathy in Alzheimer's Disease and Frontotemporal Dementia
#2 Brain Regions Involved in Arousal and Reward Processing are Associated with Apathy in Alzheimer's Disease and Frontotemporal Dementia
 |  | |||
| Lloyd A. Greene, PhD | Michael Shelanski, MD, PhD | |||
 |  |  | ||
| Jin Liu, PhD | Silvia Pasini, PhD | Carlo Corona, PhD |
This paper is part of a long-standing collaborative project with Michael Shelanski to understand the role of the transcription factor ATF4 in normal and pathologic brain function. ATF4 is an intriguing and somewhat enigmatic player in the nervous system. It is a rather unstable protein and its expression is largely regulated translationally. This allows for rapid alterations in levels, including in response to changes in the cellular environment such as activity and stress. Unlike many transcription factors, ATF4 is present in neuronal processes as well as nuclei, and is retrogradely transported from the periphery. These properties have suggested that among other things, ATF4 may function to regulate neuronal plasticity. In this context, evidence has been published variably depicting ATF4 as either a negative or a positive regulator of learning and memory. Because most of the experimental evidence in either direction has been attained by indirect means that may affect the expression or activity of multiple proteins in addition to ATF4, we have embarked on a series of studies with Drs. Jin Liu, Silvia Pasini, Carlo Corona and additional colleagues to assess the consequences of directly removing ATF4 from neurons in vitro and in vivo by means of virally-delivered shRNA constructs.
At the behavioral level, in a prior paper (Pasini et al., 2015), we reported that knockdown of ATF4 in the hippocampus of mice resulted in deficits in long-term spatial memory and behavioral flexibility without affecting associative memory. This was paralleled by impaired long-term potentiation (LTP) and long-term depression (LTD), as well as reduced glutamatergic function in tissue from both knockdown and knockout animals. At the cellular level, as reported in a previous publication (Liu et al., 2014), ATF4 knockdown in cultured cortical and hippocampal neurons and in mouse hippocampus reduced the density of dendritic "mushroom" spines, the spine-type associated with plasticity and memory. Knockdown in culture also reduced the density of excitatory synapses. Moving to the molecular level, we observed that loss of ATF4 caused a reduction in expression of both total and active Cdc42, a Rho family GTPase that among other activities, affects actin polymerization. This discovery suggested a molecular mechanism underlying the responses we observed after loss of ATF4. Indeed, ours and others’ studies showed that the various disturbances that we saw with ATF4 knockdown/knockout are phenocopied by specifically knocking down/knocking out Cdc42 in hippocampal and/or cortical neurons.
The finding that our observed effects of ATF4 loss are mediated by reduction in Cdc42 levels raised a puzzle. ATF4 is a transcription factor and rescue experiments with a transcriptionally-inactive form of the protein showed that all the cellular responses that we tested required ATF4's transcriptional activity. However, the response of Cdc42 to loss of ATF4 was not at the transcriptional level, but was due to an increase in turnover of Cdc42 protein. Thus, the question arose, which is the subject of the present Scientific Reports study, of what it is that ATF4 regulates at the transcriptional level that in turn affects Cdc42 stability? After an extensive literature search and elimination of other potential players, we focused on RhoGDIα, which had been reported to be a stabilizer of the Rho GTPases including Cdc42. We found that ATF4 down-regulation in both hippocampal and cortical neuron cultures reduces RhoGDIα protein and message levels. Moreover, chromatin immunoprecipitation (ChIP) assays revealed that that Arhgdia, the gene encoding RhoGDIα, is a direct transcriptional target of ATF4. These findings support a model (see Figure) in which ATF4 transcriptionally regulates RhoGDIα and thereby, Cdc42 levels. Therefore, by affecting Cdc42 via RhoGDIα, ATF4 modulates a variety of neuronal properties relevant to plasticity and learning, including formation of dendritic mushroom spines, stabilization of excitatory synapses, and translocation of neurotransmitter receptors to the cell surface. One additional finding reported in our paper is that the role of ATF4 in regulating actin-dependent events is not limited to neurons. With the participation of Eugenie Peze-Heidsieck, a visiting student from Agro-Paris Tech, we found that ATF4 knockdown in non-neuronal cells impairs cell migration. Because ATF4 is universally expressed, we can thus anticipate that it will have widespread actions on cell behaviors that are dependent on Cdc42 and actin polymerization.
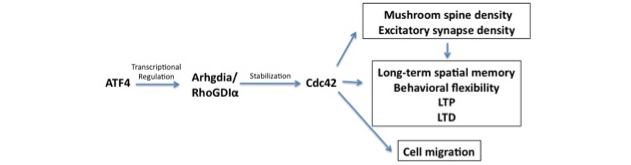 Figure: Scheme of how ATF4 regulates neuronal and cellular behavior via a RhoGDIα-Cdc42 pathway.
|
So what's on the agenda after this paper? Because we see profound effects of reducing ATF4 expression by experimental means, we are presently defining the physiologic circumstances that suppress ATF4 levels in the normal brain. With respect to the interests of the Taub Institute, given that ATF4 has been implicated in pathologic disorders such as Alzheimer’s disease and Parkinson’s disease, learning how ATF4 levels can be manipulated in neurons may provide for therapeutic advantage.
Lloyd A. Greene, PhD
Professor of Pathology and Cell Biology
lag3@cumc.columbia.edu
Michael Shelanski, MD, PhD
Henry Taub Professor of Pathology and Cell Biology
Co-Director of the Taub Institute for Research on Alzheimer's Disease and the Aging Brain
Senior Vice Dean for Research, Columbia University College of Physicians and Surgeons
mls7@cumc.columbia.edu
 |  | |
| Edward D. Huey, MD | Davangere Devanand, MBBS, MD |
Neuropsychiatric symptoms in patients with dementia are a major health problem throughout the world. These symptoms occur in the majority of dementia patients at some point during their illness, and are a major predictor of out-of-home placement and cost. A recent analysis performed in the U.K. found that agitation in patients with AD costs over 2 billion pounds annually in the U.K., with an estimated cost of over 12 billion dollars annually in the U.S. (Morris et al., 2015). The definitions of many neuropsychiatric syndromes in dementia have been “borrowed” from psychiatric syndromes developed in persons without dementia, e.g., Major Depressive Disorder, psychosis, and disinhibition. In our laboratory, we have hypothesized that the neuropsychiatric syndromes in dementia are likely dissimilar in important ways from psychiatric syndromes in neurologically-intact populations. We have attempted to redefine certain neuropsychiatric syndromes in dementia based on behavioral and emotional symptoms associated with dysfunction of specific brain regions, including arousal (Koenigs et al., 2008), dysphoria and anxiety (Huey et al., 2016), "dysexecutive" behavior (Gansler et al., 2016) (Figure 1), praxis (Huey et al., 2009), awareness of symptoms (Zamboni et al., 2010), and reward learning (Huey et al., 2015).
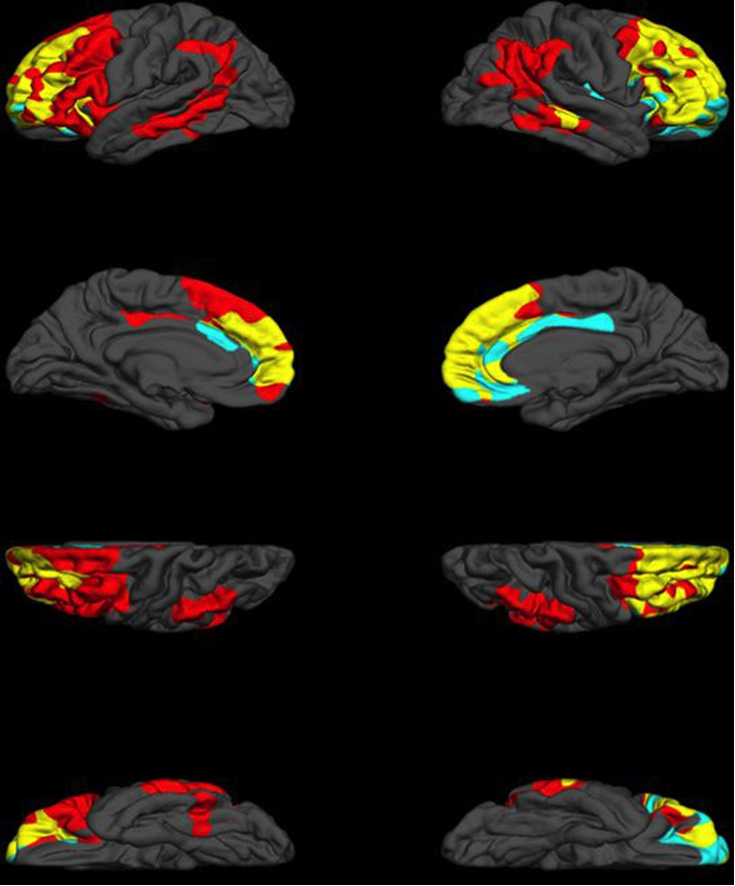
Figure 1: Neural correlates of executive function (EF) and dysexecutive behaviour (DB). Red regions indicate unique neural correlates of EF, including left premotor and supplemental motor regions, left lateral orbital cortex and bilateral middle temporal gyrus, supramarginal gyri and angular gyri. Unique neural correlates of DB are represented in teal, and include regions of the right frontal pole, right orbitofrontal cortex and bilateral anterior cingulate cortex. Finally, areas of convergence between EF and DB are in yellow, and consist of the majority of dorsolateral prefrontal cortex, frontal midline, regions and bilateral orbitofrontal cortex. Cortical thickness of the regions indicated above have been demonstrated to be significantly associated with EF or DB, respectively, and corrected for multiple comparisons. From (Gansler et al., 2016).
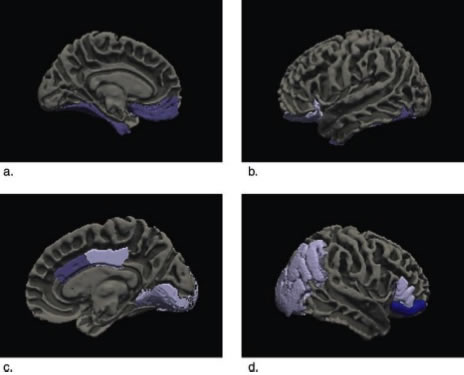
Figure 2: Significant correlations between apathy and regional volume are represented on a model brain using Freesurfer.
Apathy is an important neuropsychiatric syndrome of dementia that we have been studying recently. Apathy is defined as a deficit in motivation associated with a decrease in the performance of activities that were previously rewarding for the patient. It is a common neuropsychiatric symptom in dementia, affecting the majority of patients with AD at some point in the illness. Previous studies had linked apathy to atrophy in several different brain regions in AD, but these studies did not account for the significant co-linearity of both brain atrophy and neuropsychiatric symptoms in AD. Atrophy in one region is associated with atrophy in adjacent regions and apathy overlaps with other neuropsychiatric syndromes such as depression.
In a new study, published recently in the Journal of Alzheimer's Disease, we utilized an innovative statistical method (multivariate multiple regression with LASSO regularization) to account for co-linearity of atrophy and neuropsychiatric symptoms in 57 subjects with mild AD evaluated in the Alzheimer's Disease Neuroimaging Initiative (ADNI). Atrophy of the ventromedial prefrontal cortex including the anterior cingulate cortex, posterior cingulate and adjacent parietal and occipital cortex, and inferior temporal cortex were independently associated with apathy in AD. Previously, we had found similar areas to be associated with apathy in Frontotemporal dementia, suggesting that apathy is associated with dysfunction of the similar brain regions in different neurodegenerative disorders. These areas correspond to specific brain networks identified in non-human primates involved in arousal, threat response, and reward learning.
Edward D. Huey, MD
Associate Professor of Psychiatry and Neurology (in the Taub Institute)
edh2126@cumc.columbia.edu
Davangere Devanand, MBBS, MD
Professor of Psychiatry (in Neurology and the Gertrude H. Sergievsky Center)
dpd3@cumc.columbia.edu