
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
Best Poster Presentations
Taub Institute Retreat October 2016
White Matter Changes in Alzheimer's Disease
Sara Ebrahimi Nasrabady 1,3, Etty P. Cortes 2,3, 4, Alexander Sosunov 5, Jean-Paul G. Vonsattel 2,3,4, James E. Goldman 2, 3, Adam M. Brickman 3.
1 Division of Late Life Disorders, Department of Psychiatry, Columbia University, New York, NY
2 Department of Pathology and Cell Biology, Columbia University, New York, NY
3 Taub Institute for Research on Alzheimer's Disease and the Aging Brain, Department of Neurology, Columbia University, New York, NY
4 New York Brain Bank, College of Physicians and Surgeons, Columbia University, New York, NY
5 Department of Neurological Surgery, Columbia University, New York, NY
Sara Ebrahimi Nasrabady, MD, PhD
Alzheimer's disease (AD) is characterized by two hallmark changes in grey matter: extracellular neuritic plaques, made of amyloid-beta (Aβ), and neurofibrillary tangles, which are interneuronal aggregates of phosphorylated tau protein. However, recent neuroimaging studies show an association between white matter abnormalities and AD course and pathogenesis. For example, our research group has shown strong relationships between white matter abnormalities, which manifest as hyperintense signal (white matter hyperintensities: WMH) on T2‐weighted magnetic resonance imaging (MRI), with increased risk of incident AD, incident mild cognitive impairment (MCI), and the symptomatic course of AD. To explore the mechanistic basis of these imaging abnormalities in white matter, we studied the changes of oligodendrocyte lineage cells and the vasculature in human brain white matter in AD cases and controls. Investigation of frontal and parietal white matter revealed an increased number of Olig2 positive (Olig2+) nuclei (% total Olig2+ cells/total number of cells) in frontal deep white matter in AD compared with the controls. The measurement of Olig2+ nuclear diameter in frontal deep white matter depicts a shift toward a higher proportion of the larger nuclei in AD deep white matter tissues. Based on the differences observed in the nuclear size of oligodendrocyte lineage cells (larger nuclear size is observed in immature cells and smaller one in mature myelinating oligodendrocytes), we hypothesize that the increased number of Olig2+ cells is due to a higher number of immature oligodendrocytes in deep white matter frontal lobe. Frontal and parietal white matter vessels showed an increased level of collagen, indicated by thicker vessel walls, in AD cases compared with controls. In order to confirm that these differences were not due to age distribution between groups, we established that there was no correlation between age and small vessel wall thickness in either group. Structural changes in the white matter small vessels and the inability of oligodendrocytes to function as mature myelinating cells could be the underlying factors in the white matter damage observed in AD.
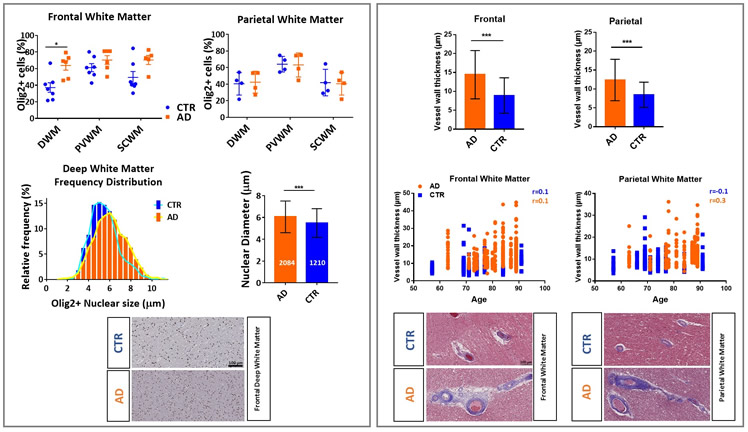 Figure: Changes of oligodendrocyte lineage cells and the vasculature in human brain white matter: Left column represents Olig2+ nuclear measurements and Olig2+ subtype changes based on the nuclear size. Right column represents the analysis of vessel wall thickness by measuring collagen layer in frontal and parietal lobes.
|
Sara Ebrahimi Nasrabady, MD, PhD
Postdoctoral Research Fellow, Taub Institute and Department of Psychiatry, Supported by NIH T32 Institutional Grant from Geriatric Psychiatry (5T32MH020004-18)
Mentors: Dr. Adam M Brickman and Dr. James E. Goldman
se2351@columbia.edu
Understanding the Role of Phosphoinositide Dysregulation in Endolysosomal Dysfunction: Implications for Alzheimer's Disease
Andre M. Miranda 1,2,3,4, Zofia M. Lasiecka 1,2, Yimeng Xu 1,2, Sanjid Shahriar 1,2, Robin B. Chan 1,2, Tiago Gil Oliveira 3,4, Gilbert Di Paolo 1,2,5.
1 Department of Pathology and Cell Biology, Columbia University Medical Center, New York, USA.
2 Taub Institute for Research on Alzheimer's Disease and the Aging Brain, Columbia University Medical Center, New York, USA
3 Life and Health Sciences Research Institute (ICVS), School of Health Sciences, University of Minho, Braga, Portugal
4 ICVS/3B's - PT Government Associate Laboratory, Braga/Guimarães, Portugal
5Denali Therapeutics, South San Francisco, California, USA
Andre Miguel Miranda
While autosomal mutations in genes encoding amyloid precursor protein (APP) and presenilins lead to rare early-onset familial cases of Alzheimer’s Disease (FAD), most cases are late-onset (LOAD) with no precise cause. Growing evidence suggests that endolysosomal and autophagic defects are key pathological features of AD, as well as other neurodegenerative disorders. Our recent lipidomic study shows a selective phosphotidylinositol-3-phosphate (PI3P) deficiency in LOAD-affected individuals as well as in FAD mouse models. PI3P is synthesized primarily by the lipid kinase VPS34 (also known as class III PI 3-kinase) and is considered a master organizer of endosomal and autophagy pathways. Mimicking this deficiency by silencing VPS34 in neurons caused endosomal enlargement and misprocessing of APP, important features of AD. However, it is still unclear how disrupting PI3P signaling impairs neuronal endosomal and lysosomal function and its relationship with altered APP metabolism. Genetic ablation of VPS34 in CAMKII-CRE;Vps34Flox/Flox mice was found to produce massive neurodegeneration at 3 months of age. Prior to neurodegeneration (at two months of age), extensive accumulation of p62 and poly-ubiquitinated proteins was observed both in the cortex and the hippocampus, suggesting severe impairment of endolysosomal and autophagic function. These findings are corroborated by the accumulation of lipids, namely cholesterol esters and sphingolipids, which are known to accumulate as a result of endolysosomal dysfunction. Pharmacological inhibition of VPS34 in neurons phenocopied autophagy impairment and lipid accumulation, and led to additional endolysosomal phenotypes, including delayed degradation and accumulation of APP COOH-terminal fragments (CTFs), in part in early endosomes. Our results further support Vps34 as a critical regulator of the endolysosomal and autophagy pathways in neurons, as well as a factor controlling the metabolism of APP and late-endosomal/lysosomal lipids.
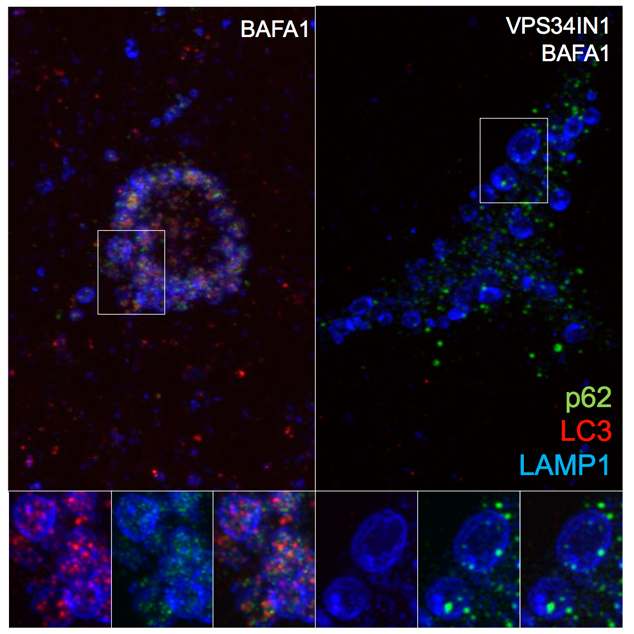 Figure: VPS34 inhibition severely affects protein clearance and causes lipid dyshomeostasis. Pharmacological inhibition of VPS34 causes accumulation of poly-ubiquitinated proteins and p62, suggesting failed clearance of autophagy-targeted substrates. Indeed, acute PI3P depletion prevents LC3 lipidation and autophagosome formation. High-resolution confocal microscope images show LC3/p62 co-localization in LAMP1+ compartment after Bafilomycin A1 treatment. VPS34 inhibition prevents assembly of LC3+ structures and causes accumulation of p62+ structures that fail to be incorporated in the lumen of late endosomes/lysosomes.
|
Andre Miguel Miranda
MD/PhD Student, School of Medicine, University of Minho and Graduate School of Arts and Sciences, Columbia University
Supported by Fundação para a Ciência e Tecnologia (PD/BD/105915/2014)
Mentors: Dr. Gilbert Di Paolo and Dr. Tiago Gil Oliveira
alm2259@columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.