
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
Featured Research
In the Lab:
Ronald K. H. Liem, PhD
 Ronald K. H. Liem, PhD
Ronald K. H. Liem, PhDThe focus of research in my laboratory is the study of the neuronal cytoskeleton and its relationship to neurodegenerative disease. A variety of neuronal intermediate filament proteins are expressed in the mammalian nervous system, and overexpression and mutations in these proteins have resulted in neurodegenerative diseases. We study the effects that these mutations have on neurofilament assembly and transport in an effort to elucidate their role in neurodegeneration.
In addition, our studies have also focused on a family of cytoskeletal linker proteins called (spectra) plakins. These proteins connect cytoskeletal elements to each other and to junctional complexes. They consist of combinations of interacting domains, which include domains that bind to microfilaments, microtubules, intermediate filaments, cell adhesion molecules, as well as members of the catenin family. Mutations in these proteins lead to a variety of cellular defects, including sensory neuron degeneration, muscular dystrophy, and axonal outgrowth defects.
Past and current projects in these primary areas of study include:
Identification of the Subunits of the Neurofilament Networks in the Central and Peripheral Nervous System (view relevant publications)
My work on neurofilaments began as a post-doc, when I first showed that neurofilaments in both the central and peripheral nervous system were composed primarily of triplet proteins: NFL, NFM, and NFH (for light, middle, and high molecular with neurofilament protein). Over the years, we have identified the components of the neurofilament triplet proteins, purified each component using biochemical methods, cloned all the complementary DNAs (cDNAs) and studied their assembly in vitro and in vivo. We have shown that the low molecular weight neurofilament (NFL) is essential for the formation of intermediate filaments consisting of the neurofilament triplet proteins.
Subsequently, we identified a new component of neuronal intermediate filaments, α−internexin, as part of the neurofilamentous network. We have isolated and sequenced cDNA and genomic clones of α−internexin and studied the developmental regulation of the neuronal intermediate filaments in the nervous system. We have shown that α−internexin can self-assemble and is expressed earlier in development than the neurofilament triplet proteins. We have also studied the regulation of expression of α−internexin in the nervous system and shown that overexpression will result in the degeneration of Purkinje cells in the cerebellum.
Abnormal Accumulations of Neuronal Intermediate Filaments (view relevant publications)
Abnormal neurofilamentous accumulations have long been described as being present in neuronal processes and cell bodies in various neurodegenerative diseases. A clear link between mutations in the gene for the neurofilament light protein (NEFL) and neurodegenerative diseases has been established in Charcot-Marie-Tooth disease, where as many as fourteen dominant mutations have been identified. These dominant mutations in the NFL protein result in the formation of neurofilamentous aggregates that cause abnormalities in mitochondrial and slow axonal transport, as well as neuritic degeneration. In cell transfection studies, we have shown a correlation between the presence of pathogenic mutations in NEFL and abnormal assembly of the filaments in both neuronal and non-neuronal cells. In a collaborative project with Dr. Howard Worman and Dr. Victor Lin, we generated a knock-in mouse model with one of the mutations, N98S. This NeflN98S/+ mouse recapitulates most of the features of patients with this mutation. The animals had a tremor and showed decrease in balance. Immunohistochemical analyses showed multiple inclusions in the cell bodies and axons of spinal cord neurons and dorsal root ganglia (Figure 1), disorganized processes in the cerebellum, and abnormal processes in the cerebral cortex and pons. Abnormal processes were observed as early as post-natal day 7. Electron microscopic analysis of sciatic nerves showed a reduction in the number of neurofilaments, an increase in the number of microtubules and a decrease in the axonal diameters.
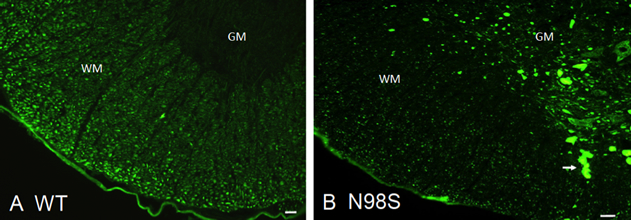 Figure 1. Immunofluorescence micrographs of sections of spinal cord stained with anti-NFL-N antibody showing inclusions in NeflN98S/+ mice. (A) Section of anterior horn of 18-month old A. Nefl+/+ (wild-type: WT) mouse, and (B) NeflN98S/+ (N98S) mouse. WM (white matter) of Nefl+/+ mice shows axonal labeling, whereas there is relatively little labeling in the cell bodies of the GM (gray matter). In contrast, there was relatively little labeling in the WM of NeflN98S/+ mice, except for the inclusions (arrow), which are likely due to axonal swellings. Adapted from Adebola et al. 2015, Fig. 3.
|
We conducted ultrastructural analyses of cultured dorsal root ganglia (DRG) from NeflN98S/+ mice and showed that inclusions found in DRG soma and neurites can occur in embryonic stages. These studies also revealed that the inclusions are disordered neurofilaments packed in high density, segregated from other organelles (Figure 2).
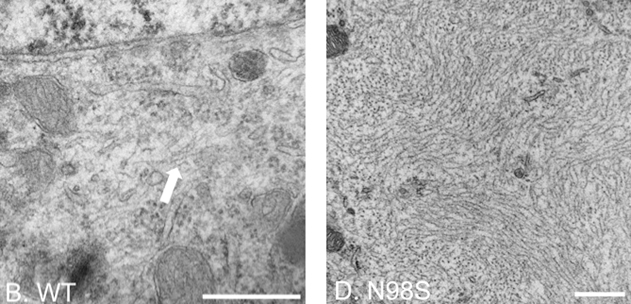 Figure 2. Electron micrographs of DRG cell soma of wild-type and NeflN98S/+ mice. The filamentous structure formed by intermediate filaments is pointed by the arrow in Nefl+/+ DRG (B). Massive accumulation of disordered neurofilaments is observed in NeflN98S/+ DRG (D). The density of neurofilaments is very high and few other cytoplasmic elements can be seen within the aggregates. Scale bars = 500 nm. From Zhao et al. 2017, Fig. 3.
|
This NeflN98S/+ mouse provides an excellent model to study the pathogenesis of the neurodegeneration observed in human Charcot-Marie-Tooth type 2, and should prove useful for testing potential therapies. Further studies using motor neurons derived from the ES cells that were used to generate the NeflN98S/+ mice, as well as cultured dorsal root ganglia from these mice, are currently underway and are supported by a grant from the Charcot-Marie-Tooth Association.
Studies on the cytoskeletal linker proteins, MACF1 and BPAG1 (view relevant publications)
In our search for intermediate filament associated proteins in the nervous system, we characterized the isoforms of BPAG1 (bullous pemphigoid antigen 1) in greater detail. BPAG1 had a known isoform that links the intermediate filaments to the hemidesmosomes in epidermal cells (BPAG1e). BPAG1e is a member of the plakin family of proteins that contain what was thought to be a unique plakin domain, and that linked intermediate filaments to junctional complexes. Interestingly, knock-outs of the BPAG1 gene resulted in only minor skin abnormalities, but the mice had severe sensory neuron degeneration. The BPAG1 knock-out mouse was also found to be identical to a known mouse mutant, called dystonia musculorum (dt). We investigated in more detail the gene structure and alternative splicing of the BPAG1 gene and found that there were multiple isoforms with different functional units of the BPAG1 gene, including one in the nervous system that we called BPAG1a, which has microtubule and actin binding domains and a series of spectrin repeats serving as a spacer (Figure 3). The removal of this isoform in mice was the cause for the sensory neuron defects. This isoform has no intermediate filament binding domain.
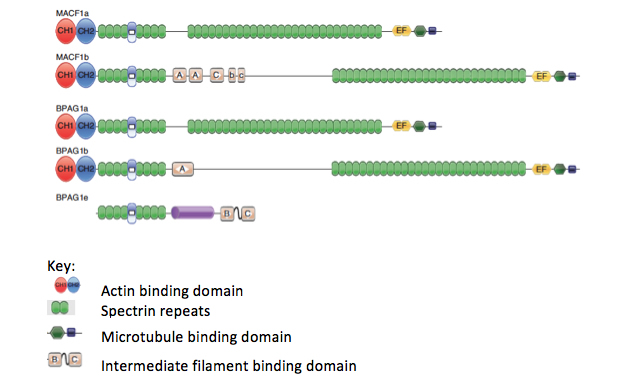 Figure 3. Diagrams of the different splice variants of BPAG1 and MACF1 showing the domains that can interact with cytoskeletal elements, including actin, microtubules, and intermediate filaments. The spectrin repeat domains interact with other cellular elements, including junctional proteins. Adapted from Liem, 2016, Fig. 1.
|
During this time, we also identified a related cytoskeletal linker protein that can interact with microtubules and actin filaments, which we called MACF1 (microtubule actin cross-linking factor 1). This protein has an N-terminal actin binding motif that is functional in vitro and in vivo and a novel C-terminal microtubule binding motif. In addition, the protein has a series of dystrophin-like spectrin repeats. Complete knock-out of the protein results in an early embryonic lethal phenotype that was similar to a Wnt3-/- animal, and our results suggest a role of MACF1 in the wnt signaling pathway. By doing a nervous system specific knock-out, we showed that MACF1 plays a critical role in neuronal migration. BPAG1 and MACF1 are closely related in domain structure to the MACF1 molecule, but the two proteins appear to have different functions, since the knock-outs have different phenotypes. Structural analysis also showed that the plakin domain is actually composed of spectrin repeats and that the plakins are members of the spectrin superfamily of proteins.
Ronald K. H. Liem, PhD
Professor of Pathology and Cell Biology (in the Taub Institute)
rkl2@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.